Objective: To examine the effectiveness of switching patients to lurasidone using 3 different dosing strategies.
Method: Adults with DSM-IV-defined schizophrenia or schizoaffective disorder in a nonacute phase of illness were randomized to 1 of 3 lurasidone dosing regimens for the initial 2 weeks of the study: (1) 40 mg/d for 2 weeks; (2) 40 mg/d for 1 week, increased to 80 mg/d on day 8 for week 2 (up-titration group); and (3) 80 mg/d for 2 weeks. Lurasidone was then flexibly dosed (40-120 mg/d) for the subsequent 4 weeks of the study. The preswitch antipsychotic agent was tapered by day 7 to 50% of the original dose and discontinued by the end of week 2. Subjects were stratified on the basis of whether the primary preswitch antipsychotic medication was classified as “sedating” (olanzapine or quetiapine) or “nonsedating” (all other antipsychotics). The primary outcome measure was time to treatment failure, defined as any occurrence of insufficient clinical response, exacerbation of underlying disease, or discontinuation due to an adverse event. The study was conducted from June 2010 to May 2011.
Results: Of 240 subjects treated in this study, 86 (35.8%) were treated with an antecedent sedating antipsychotic, and 154 (64.2%) were treated with an antecedent nonsedating antipsychotic. Nineteen (7.9%) of the 240 patients experienced treatment failure. No clinically relevant differences were observed when the 3 randomized switch groups were compared. Treatment failure rates were 10/86 (11.6%) versus 9/154 (5.8%) among subjects who had been receiving a preswitch sedating versus nonsedating antipsychotic medication, respectively. Consistent with prior studies of lurasidone, there was no signal for clinically relevant adverse changes in body weight, glucose, insulin, lipids, or prolactin; mean improvements in weight and lipids were observed. Movement disorder rating scales did not demonstrate meaningful changes. The incidence of akathisia as an adverse event was 12.5%; only 1 subject (0.4%) discontinued due to akathisia.
Conclusions: Switching patients to lurasidone can be successfully accomplished by starting at 40 mg/d for 2 weeks, or 80 mg/d for 2 weeks, or 40 mg/d for 1 week followed by 80 mg/d the second week.
Trial Registration: ClinicalTrials.gov identifier: NCT01143077
J Clin Psychiatrist
© Copyright 2013 Physicians Postgraduate Press, Inc.
Submitted: July 1, 2012; accepted December 12, 2012.
Online ahead of print: January 15, 2013 (doi:10.4088/JCP.12m07992).
Corresponding author: Leslie Citrome, MD, MPH, 11 Medical Park Dr, Ste 106, Pomona, NY 10970 ([email protected]).
Effectiveness of Lurasidone in Patients with Schizophrenia or Schizoaffective Disorder Switched From Other Antipsychotics:
A Randomized, 6-Week, Open-Label Study
ABSTRACT
Objective: To examine the effectiveness of switching patients to lurasidone using 3 different dosing strategies.
Method: Adults with DSM-IV–defined schizophrenia or schizoaffective disorder in a nonacute phase of illness were randomized to 1 of 3 lurasidone dosing regimens for the initial 2 weeks of the study: (1) 40 mg/d for 2 weeks; (2) 40 mg/d for 1 week, increased to 80 mg/d on day 8 for week 2 (up-titration group); and (3) 80 mg/d for 2 weeks. Lurasidone was then flexibly dosed (40–120 mg/d) for the subsequent 4 weeks
of the study. The preswitch antipsychotic agent was tapered by day 7 to 50% of the original dose and discontinued by the end of week 2. Subjects were stratified on the basis of whether the primary preswitch antipsychotic medication was classified as “sedating” (olanzapine or quetiapine) or “nonsedating” (all other antipsychotics). The primary
outcome measure was time to treatment failure, defined as any occurrence of insufficient clinical response, exacerbation of underlying disease, or discontinuation due to an adverse event. The study was conducted from June 2010 to May 2011.
Results: Of 240 subjects treated in this study, 86 (35.8%) were treated with an antecedent sedating antipsychotic, and 154 (64.2%) were treated with an antecedent nonsedating antipsychotic. Nineteen (7.9%) of the 240 patients experienced treatment failure. No clinically relevant differences were observed when the 3 randomized switch groups were compared. Treatment failure rates were 10/86 (11.6%) versus 9/154 (5.8%) among subjects who had been receiving a preswitch sedating versus nonsedating antipsychotic medication, respectively. Consistent with
prior studies of lurasidone, there was no signal for clinically relevant adverse changes in body weight, glucose, insulin, lipids, or prolactin; mean improvements in weight and
lipids were observed. Movement disorder rating scales
did not demonstrate meaningful changes. The incidence
of akathisia as an adverse event was 12.5%; only
1 subject (0.4%) discontinued due to akathisia.
Conclusions: Switching patients to lurasidone can be successfully accomplished by starting at 40 mg/d for 2 weeks, or 80 mg/d for 2 weeks, or 40 mg/d for 1 week followed by
80 mg/d the second week.
Trial Registration: ClinicalTrials.gov identifier: NCT01143077
J Clin Psychiatrist
© Copyright 2013 Physicians Postgraduate Press, Inc.
Submitted: July 1, 2012; accepted December 12, 2012.
Online ahead of print: January 15, 2013 (doi:10.4088/JCP.12m07992).
Corresponding author: Leslie Citrome, MD, MPH, 11 Medical Park Dr,
Ste 106, Pomona, NY 10970 ([email protected]).
Lurasidone is a second-generation antipsychotic that received approval in October 2010 by the US Food and Drug Administration for the treatment of schizophrenia.1 The recommended starting dose is 40 mg/d, and the maximum recommended dose is 160 mg/d.1 Regulatory approval of lurasidone was supported by results from 5 positive, 6-week, randomized, placebo-controlled trials that demonstrated the efficacy of lurasidone at fixed doses ranging from 40–160 mg administered once daily1; primary reports for 3 of the studies have been published to date,2–4 in addition to a pooled analysis of all 5 studies using the metric of number needed to treat.5 Lurasidone can be differentiated from other available second-generation antipsychotics by its receptor binding profile, with high affinities for the serotonin 5-HT7, norepinephrine α2C (antagonist), and serotonin 5-HT1A (weak-moderate partial agonist) receptors in addition to the expected high binding affinity for dopamine D2 and serotonin 5-HT2A receptors. Lurasidone has little to no appreciable affinity for 5-HT2C, histamine H1, and acetylcholine M1 receptors. Lurasidone is associated with minimal weight gain and no clinically meaningful alterations in glucose, lipids, prolactin, or the electrocardiogram QT interval.1
The management of patients with schizophrenia is complex, and there is marked heterogeneity in treatment response. An antipsychotic medication that is efficacious and tolerable for one person can be inadequate and unacceptable for another. Medications themselves display differences in terms of efficacy profiles and propensity for different adverse effects. Matching up the best medication for the individual patient is an empirical decision and can be a substantial challenge.6 Thus, switching between antipsychotic medications commonly occurs in the routine treatment of schizophrenia in an effort to find the optimal regimen for an individual patient.7,8 A number of studies have examined outcomes of antipsychotic medication switches, with perhaps the largest and best-known being the National Institute of Mental Health–funded Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE), in which approximately 1,500 patients with schizophrenia were enrolled; depending on the circumstances or reason for the switch and the medication the patient was switched from, different outcomes were observed for the antipsychotics tested.9 Switching for the purpose of assessment of improvement in metabolic variables was the specific focus of a recently reported study of aripiprazole,10 with benefits and risks observed similar to those shown in switch studies involving ziprasidone.11
To better understand the effects of switching antipsychotic medication regimens to lurasidone among outpatients under “real-world” conditions, we undertook this study to evaluate the effectiveness of this switch in patients with schizophrenia or schizoaffective disorder. We examined if patients could be successfully switched to lurasidone 40 or 80 mg/d over an initial 2-week period during which their prior antipsychotic was simultaneously decreased. We examined if there were any differences in effectiveness over the 6-week study period between the strategies of initially switching to
40 mg/d, initially switching to 80 mg/d, or titrating in a stepwise progression from 40 to 80 mg/d.
METHOD
This multicenter, randomized (to 1 of 3 initial titration schedules), open-label, parallel-group 6-week study was conducted at 28 sites in the United States (ClinicalTrials.gov identifier: NCT01143077). The study was reviewed and approved by an institutional review board at each
study center, and the trial was conducted in accordance with Good Clinical Practice as required by the International Conference on Harmonization guidelines. Compliance with these requirements also constitutes conformity with the
ethical principles of the Declaration of Helsinki.
Participants
Inclusion criteria included age ≥ 18 years, fulfillment of DSM-IV criteria for a primary diagnosis of schizophrenia or schizoaffective disorder established by semistructured interview, duration of illness ≥ 1 year, considered to be an appropriate candidate for switching current antipsychotic medication due to insufficient efficacy and/or safety or tolerability concerns, “clinically stable” (nonacute phase of illness) for at least 8 weeks prior to baseline as defined by Clinical Global Impressions-Severity of Illness scale (CGI-S)12 score ≤ 4 (at both screening and baseline), dose of the preswitch antipsychotic(s) was stable (± 50%) for at least 28 days prior to screening (up to protocol specified maximum dose), and no exacerbation of schizophrenia or schizoaffective disorder had occurred for at least 8 weeks prior to screening. Subjects taking 2 antipsychotic medications (but not more) at screening were eligible for study inclusion, but treatment with the antipsychotic medication determined to be “secondary,” on the basis of investigator judgment, was to be discontinued prior to randomization, and all subjects were to be receiving a single antipsychotic medication at randomization. Subjects were required to either be outpatients or be currently receiving treatment in a nonacute long-term inpatient setting.
Exclusion criteria included presence of an Axis I or Axis II disorder other than schizophrenia or schizoaffective disorder that was the primary focus of treatment prior to screening, or total daily dose of preswitch antipsychotic medication exceeding the following during the 28 days prior to screening: aripiprazole 30 mg, asenapine 20 mg, iloperidone 24 mg, olanzapine 20 mg, paliperidone 12 mg, quetiapine 800 mg, risperidone 8 mg, or ziprasidone 160 mg; in the case of first-generation antipsychotics, the dose must not have exceeded the equivalent of haloperidol 12 mg/d. Also excluded were subjects who experienced persistent lack of improvement in psychotic symptoms despite adequate trials (at least 6 weeks at standard doses) of 2 or more antipsychotic agents in the 12 months prior to screening; subjects considered by the investigator to be at imminent risk of suicide or harm to self, others, or property; and subjects who had suicidal ideation at baseline or had attempted suicide within 90 days prior to randomization.
Interventions
All subjects underwent a screening period of up to
14 days during which time they continued receiving their preswitch antipsychotic medication. Subjects who continued to meet entry criteria were randomly assigned to 1 of 3 open-label lurasidone arms: (1) lurasidone 40 mg/d for 14 days, followed by flexible dosing between 40 and 120 mg/d for 4 weeks; (2) lurasidone 40 mg/d for 7 days, then 80 mg/d for 7 days, followed by flexible dosing between 40
and 120 mg/d for 4 weeks; and (3) lurasidone 80 mg/d for 14 days, followed by flexible dosing between 40 and 120 mg/d for 4 weeks. The subject’s preswitch antipsychotic dose was reduced by 50% by day 7, followed by complete discontinuation by day 14. At randomization, via an Interactive Voice Response System, subjects were stratified on the basis of whether the preswitch antipsychotic medication was “sedating” (olanzapine or quetiapine) or “nonsedating” (all other antipsychotics). Stratification was prespecified per protocol as part of the randomization scheme to ensure an equal distribution of “sedating” and “nonsedating” preswitch medications across the 3 treatment arms. Lurasidone was administered once daily in the evening, with food or within 30 minutes after eating.
Treatment with benztropine (up to 6 mg/d) was permitted as needed for extrapyramidal symptoms. In cases in which benztropine was not available or a subject had an inadequate response or intolerability to benztropine treatment, biperiden (up to 16 mg/d), trihexyphenidyl (up to 15 mg/d), or diphenhydramine (up to 100 mg/d) could be used to treat acute extrapyramidal symptoms. Treatment with propranolol (up to 120 mg/d) was permitted as needed for akathisia. Medications used to treat movement disorders could not be given prophylactically.
Concomitant use of lorazepam, temazepam, or zolpidem was permitted during the study at the discretion of the investigator with the following restrictions: lorazepam was permitted up to 4 mg/d for anxiety symptoms or agitation, as clinically indicated. Zolpidem (≤ 10 mg/d), zolpidem extended-release (≤ 12.5 mg/d), temazepam (≤ 30 mg/d), eszopiclone (≤ 3 mg/d), and zaleplon (≤ 10 mg/d) could be administered at bedtime for insomnia, as needed. Hypnotic agents were to be administered no more than once nightly. Subjects could be treated with any marketed mood stabilizers (eg, lithium, divalproex, and lamotrigine) or antidepressants during the course of the study, at the discretion of the investigator. Potent inducers or inhibitors of the CYP3A4 enzyme system were prohibited during all phases of this study, as was the use of herbal supplements (eg, Ginkgo biloba, kava, and St John’s wort) or other complementary or alternative agents. Ongoing psychotherapeutic and psychosocial interventions were permitted during the course of this trial.
Outcomes
The primary outcome (effectiveness) was time to treatment failure, defined as any occurrence of insufficient clinical response, exacerbation of underlying disease, or discontinuation due to an adverse event (AE), as determined by investigator judgment. Secondary outcomes included time to discontinuation for any reason (all-cause discontinuation); incidence of AEs; and change from baseline to week 6 endpoint in weight, body mass index, waist circumference, fasting lipids, glucose, hemoglobin A1c, insulin, C-reactive protein, prolactin, and scores on the following scales: Abnormal Involuntary Movement Scale (AIMS),13 Barnes Akathisia Scale (BAS),14 Simpson-Angus Scale (SAS),15 Positive and Negative Syndrome Scale (PANSS),16 CGI-S,12 Calgary Depression Scale for Schizophrenia (CDSS),17 and Columbia Suicide Severity Rating Scale.18 Secondary outcomes that will be reported in a separate publication included the Personal Evaluation of Transitions in Treatment (PETiT),19 Health Services Utilization Questionnaire,20 Short Form–12 Health Survey,21 and Medication Satisfaction Questionnaire.22
Statistical Analysis
The sample size was determined by the width of the 95% confidence interval for the proportion of subjects within each lurasidone group expected to experience “treatment failure” by week 6 to ensure the precision of the estimate of the true treatment failure rate. Assuming (1) 20% of lurasidone-treated subjects would experience “treatment failure” by week 6 and (2) 9% as the half width (the distance between the upper/lower limits to the point estimate) of the 95% confidence interval (CI; ie, 11%–29%), approximately 80 subjects per group would be required. In addition, the 95% CI for the treatment failure rate of the overall sample of 240 subjects would be 15%–25%.
Any subject who received at least 1 dose of lurasidone was included in the safety population (study population). All effectiveness and safety analyses were performed on the safety population. The intent-to-treat (ITT) population was defined as all subjects who were randomized, received at least 1 dose of study drug, and had a baseline and at least 1 postbaseline efficacy measurement, all from the same scale. The ITT population was used for the efficacy analyses. The only statistical inferences were in time to treatment failure or all-cause discontinuation and within-treatment change in efficacy scales, so there were no multiplicity considerations. No imputation was made for missing values. P value threshold for significance was .05.
Time to treatment failure and time to discontinuation for any reason were summarized using Kaplan-Meier plots (survival analysis) and descriptive statistics. Subjects whose preswitch antipsychotic medication was classified as “sedating” (olanzapine or quetiapine) or “nonsedating” (all other antipsychotics) were further contrasted using treatment failure rates and descriptively using number needed to harm (NNH). The AE analyses included the proportion of subjects with discontinuation due to AE. Descriptive statistics for continuous and discrete safety variables as well as shift tables were compiled and are presented as appropriate. Least squares means and their respective 95% CIs for the efficacy outcomes were calculated from an analysis of covariance (ANCOVA) with treatment and pooled center as fixed factors and baseline value as a covariate.
The software used for all analyses and summaries was SAS Version 9.2 (SAS Institute; Cary, North Carolina).
RESULTS
A total of 377 subjects provided informed consent and were screened to participate in the study, of whom 244 (64.7%) were randomized. The first subject was enrolled on June 24, 2010, and the last subject completed the study on May 19, 2011. Of the 244 subjects who were randomized to receive study medication, 74 were assigned to lurasidone 40 mg/d in weeks 1 and 2; 88, to receive 40 mg/d in week 1 followed by 80 mg/d in week 2; and 82, to receive 80 mg/d in weeks 1 and 2. A total of 4 subjects were randomized and did not receive study drug. Of these subjects, 3 subjects withdrew consent, and 1 subject was hospitalized prior to the first dose. Figure 1 provides a description of subject disposition. Table 1 shows subject demographics and baseline clinical characteristics of the study population. In the study population, 86/240 (35.8%) were treated with an antecedent sedating antipsychotic medication (olanzapine or quetiapine), and 154/240 (64.2%) were treated with an antecedent nonsedating antipsychotic.
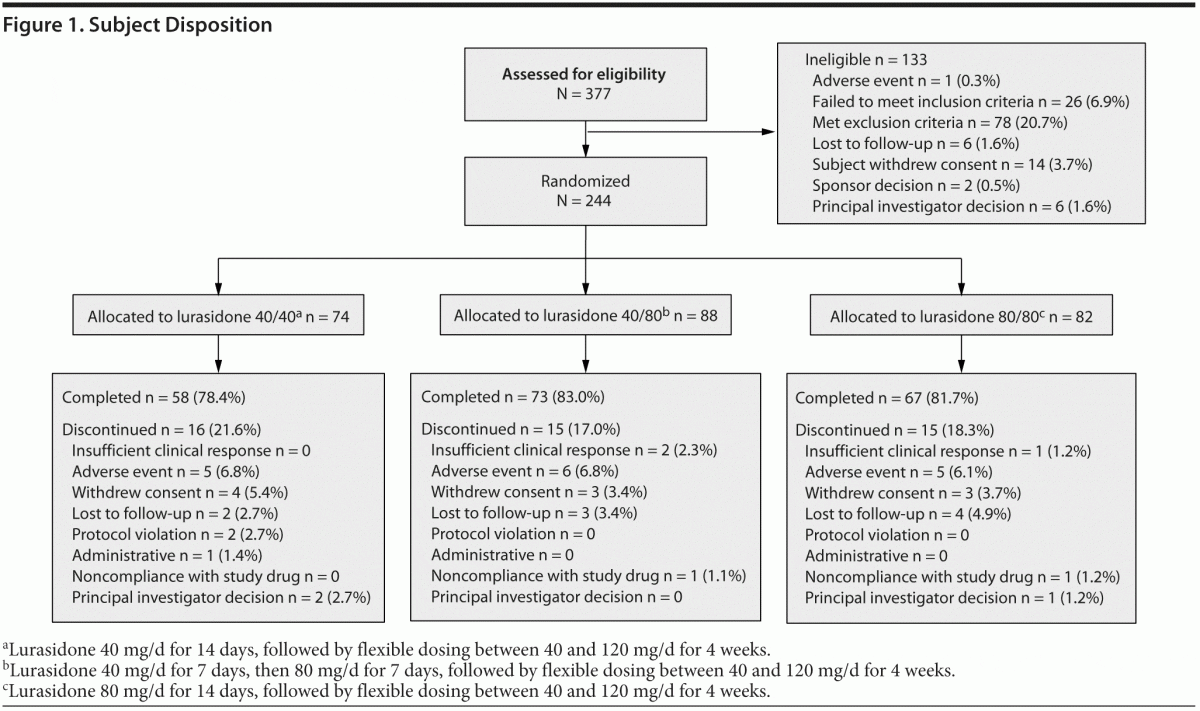
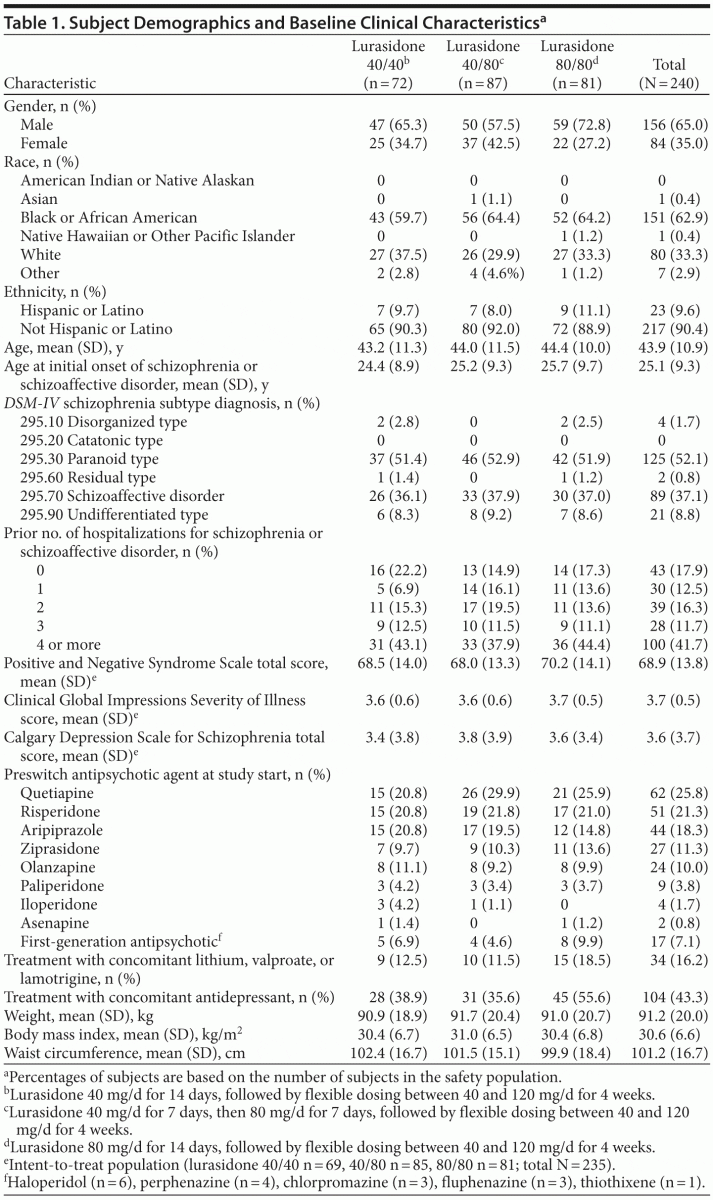
Lurasidone 40, 80, and 120 mg/d were the modal daily doses for 51 (21.3%), 119 (49.6%), and 70 (29.2%) of the subjects, respectively. The proportion of randomized subjects who completed 2 weeks and who were receiving lurasidone antipsychotic monotherapy free of concomitant medication at study endpoint was 192/224 (85.7%).
Effectiveness Outcomes
The primary outcome, time to treatment failure, is described in Table 2 and Figure 2. No statistically significant or clinically relevant differences in time to treatment failure were observed among the 3 randomized treatment groups (log-rank P = .861). A total of 19 subjects (7.9%) experienced treatment failure (based on the prespecified definition of treatment failure), and a total of 42 subjects (17.5%) discontinued study treatment overall.
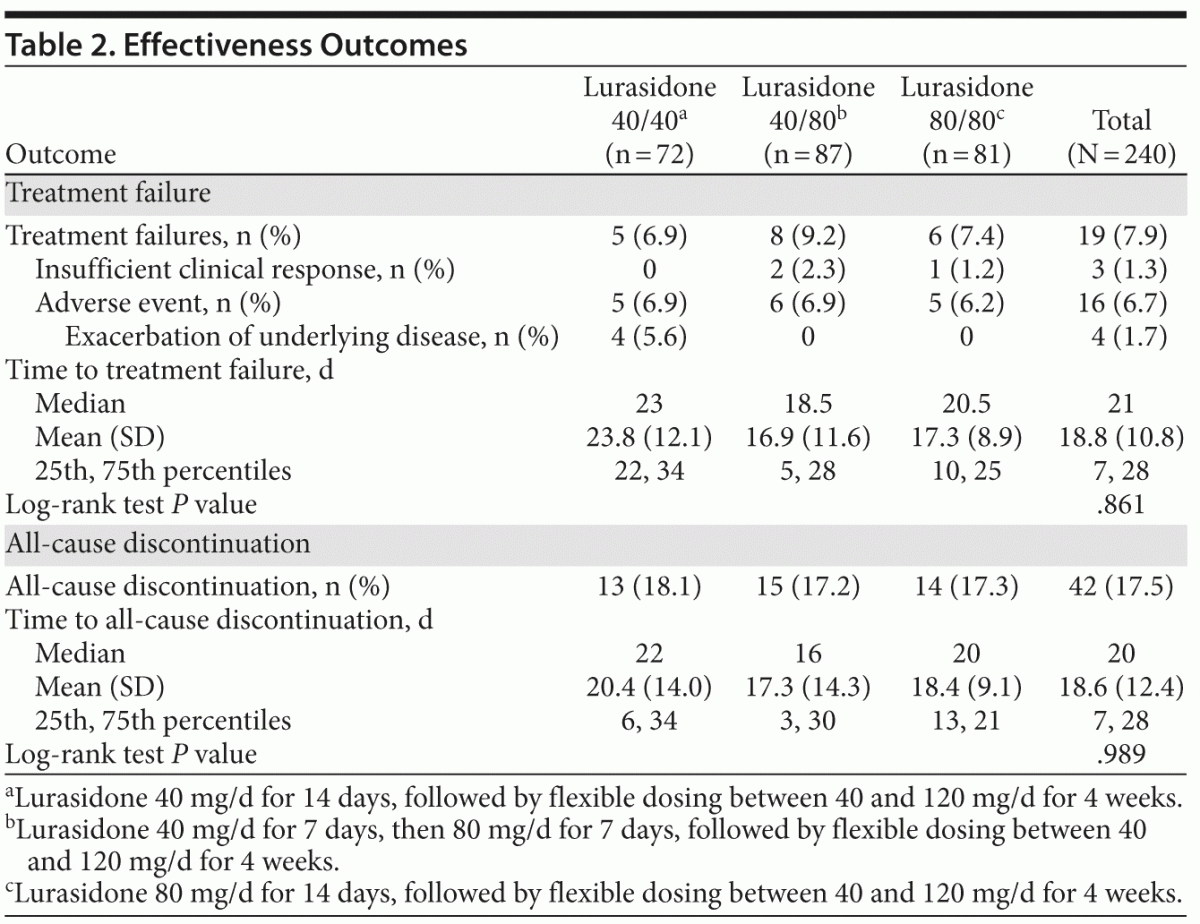
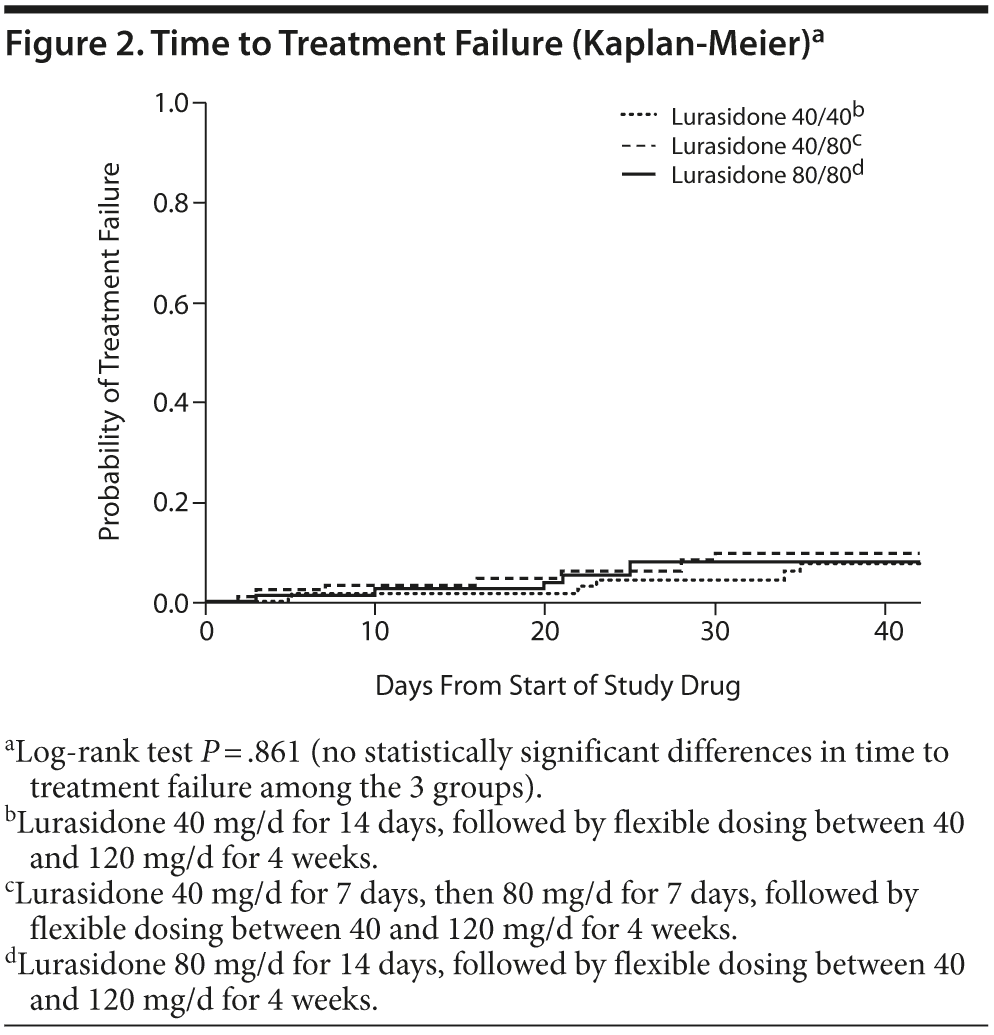
Differences between strata. Differences emerged between subjects who had been receiving a sedating antipsychotic (olanzapine or quetiapine) immediately prior to the switch to lurasidone versus those who were receiving a nonsedating antipsychotic (all others). Time to treatment failure differed numerically (log-rank P = .1008) among subjects who had been receiving a sedating antipsychotic compared to those who were receiving a nonsedating antipsychotic (see Supplementary eFigure 1 at PSYCHIATRIST.COM); treatment failure rates were 10/86 (11.6%) versus 9/154 (5.8%), respectively. The secondary outcome of time to discontinuation for any reason is described in Table 2 (see also Supplementary eFigure 2). Time to all-cause discontinuation differed significantly (log-rank P = .0368) for those receiving a sedating antipsychotic compared to those receiving a nonsedating antipsychotic. Discontinuation rates were 21/86 (24.4%) for subjects who had been receiving a sedating antipsychotic immediately prior to the switch (6/23 [26.1%] of subjects receiving quetiapine at baseline and 15/63 [23.8%] of subjects receiving olanzapine at baseline) versus 21/154 (13.6%) for subjects who had been receiving a nonsedating antipsychotic, resulting in a NNH of 10 (95% CI, 5–482) in favor of the nonsedating group.
Safety and Tolerability Outcomes
The incidences of the most commonly encountered AEs, as defined by frequency ≥ 5% among all patients, are noted in Supplementary eTable 1. Among all subjects, the rates of treatment-emergent nausea, insomnia, akathisia, headache, vomiting, somnolence, and dry mouth were 13.8%, 12.9%, 12.5%, 9.6%, 7.1%, 6.7%, and 5.8%, respectively. Differences in rates were observed between subjects who were previously treated with a sedating antipsychotic versus those who received a nonsedating antipsychotic. For example, among all lurasidone treatment groups, for the sedating group and nonsedating group, respectively, insomnia was found for 16/86 (18.6%) vs 15/154 (9.7%) of the subjects; fatigue, 7/86 (8.1%) vs 3/154 (1.9%); vomiting, 8/86 (9.3%) vs 9/154 (5.8%); somnolence, 3/86 (3.5%) vs 13/154 (8.4%); sedation, 2/86 (2.3%) vs 8/154 (5.2%); akathisia, 12/86 (14%) vs 18/154 (11.7%); and anxiety, 5/86 (5.8%) vs 4/154 (2.6%). Discontinuation because of an AE was observed in 16/240 (6.7%) of the entire sample, but was evidenced in 10/86 (11.6%) vs 6/154 (3.9%) for subjects who had received an antecedent sedating antipsychotic versus a nonsedating antipsychotic. This difference was statistically significant, with a NNH of 13 (95% CI, 7–335) in favor of the nonsedating group. Of the subjects who discontinued because of an AE, exacerbation of underlying disease (n = 2), insomnia (n = 3), anxiety (n = 1), akathisia (n = 1), nausea (n = 1), upper abdominal pain (n = 1), and vomiting (n = 1) were observed in the sedating group; exacerbation of underlying disease (n = 2), somnolence (n = 1), muscular weakness (n = 1), dysphoria (n = 1), and agitation (n = 1) were observed in the nonsedating group.
Treatment-emergent serious adverse events (SAEs) were uncommon (5 subjects, 2.1%); of the 5 subjects with SAEs, 3 subjects (4.2%) were in the lurasidone 40 mg/40 mg treatment group, and 2 subjects (2.3%) were in the lurasidone 40 mg/80 mg treatment group. There were no subjects with SAEs in the lurasidone 80 mg/80 mg treatment group. Two subjects (0.8%) had a schizoaffective disorder reported as a treatment-emergent SAE. One subject each (0.4%) had osteoarthritis, alcoholism, schizophrenia, and sexually inappropriate behavior reported as treatment-emergent SAEs. Treatment-emergent SAEs were reported in 1 subject (1.2%) in the sedating stratum and 4 subjects (2.6%) in the nonsedating stratum. There were no subject deaths during the study.
The changes from baseline to last-observation-carried-forward (LOCF) endpoint in weight, metabolic variables, and prolactin are listed in Supplementary eTable 2. There was no signal for clinically relevant changes on any of these variables. (Total numbers of patients vary because postbaseline determinations were not available for all subjects for all measures.) Among all randomized subjects in the safety population, 2/220 (0.9%) experienced weight gain ≥ 7% from baseline; 4/220 (1.8%) experienced weight loss ≥ 7% from baseline. No subject had a total cholesterol value ≥ 300 mg/dL or low-density lipoprotein cholesterol ≥ 200 mg/dL. Levels of triglycerides ≥ 300 mg/dL were observed in 5/193 (2.6%) of subjects. Glucose > 160 mg/dL was observed in 1/194 (0.5%) of the subjects, and hemoglobin A1c ≥ 7.5% was observed in 1/218 (0.5%) of the subjects. Prolactin ≥ 5 × upper limit of normal was observed in 2/219 (0.9%) of subjects, all in the randomized group that initially received lurasidone 80 mg/d for 14 days. See also Supplementary eTable 3.
Median and mean SAS, BAS, and AIMS scores did not demonstrate meaningful changes; median change from baseline to LOCF endpoint on these 3 scales was 0, and mean change on the SAS was 0, regardless of how patients were randomized to start lurasidone treatment. Mean change on the BAS was −0.1 and on the AIMS was 0 for the study population (see also Supplementary eTable 4). With regard to akathisia, at LOCF endpoint 205/231 (88.7%) of subjects had a BAS score rated as “absent,” 9/231 (3.9%) had a rating of “questionable,” 14/231 (6.1%) had a rating of “mild,” 3/231 (1.3%) had a rating of “moderate,” and none had a rating of “marked” or “severe.” SAS scores rated as “abnormal” or “normal” at LOCF endpoint and BAS global assessment scores and AIMS global severity scores that worsened, improved, or remained unchanged are summarized in Supplementary eTable 5. Among all subjects, 93.1% experienced an unchanged or improved score on BAS global assessment; 95.7%, an unchanged or improved AIMS global severity score; and 96.5%, a normal SAS mean score. Treatment with anticholinergic medication (not including diphenhydramine when specifically used for insomnia or allergies) was observed in 43 subjects (17.9%). Of the 43 subjects receiving anticholinergic medication, 11 (4.6%) received these for a specific indication of akathisia. β-Adrenergic blockers (propranolol) were used specifically for akathisia in
3 subjects (1.3%).
Concomitant use of benzodiazepines varied within the range of 38 to 41 subjects (15.8%–17.1%) over the 6 weeks of the study. Among the patients who had received a sedating preswitch antipsychotic, the use of benzodiazepines ranged from 12 to 14 subjects (14.0%–16.3%). Among the patients who had received a nonsedating preswitch antipsychotic, the use of benzodiazepines ranged from 26 to 28 subjects (16.9%–18.2%). Concomitant use of zolpidem (the most commonly used hypnotic in this study) was observed in
44 subjects (18.3%).
Columbia Suicide Severity Rating Scale outcomes at LOCF endpoint showed 2/235 subjects (0.9%) with emergence of suicidal ideation (none with serious suicidal ideation), 2/235 (0.9%) with worsening of suicidal ideation, and 0/235 with emergence of suicidal behavior or attempt.
Efficacy Outcomes
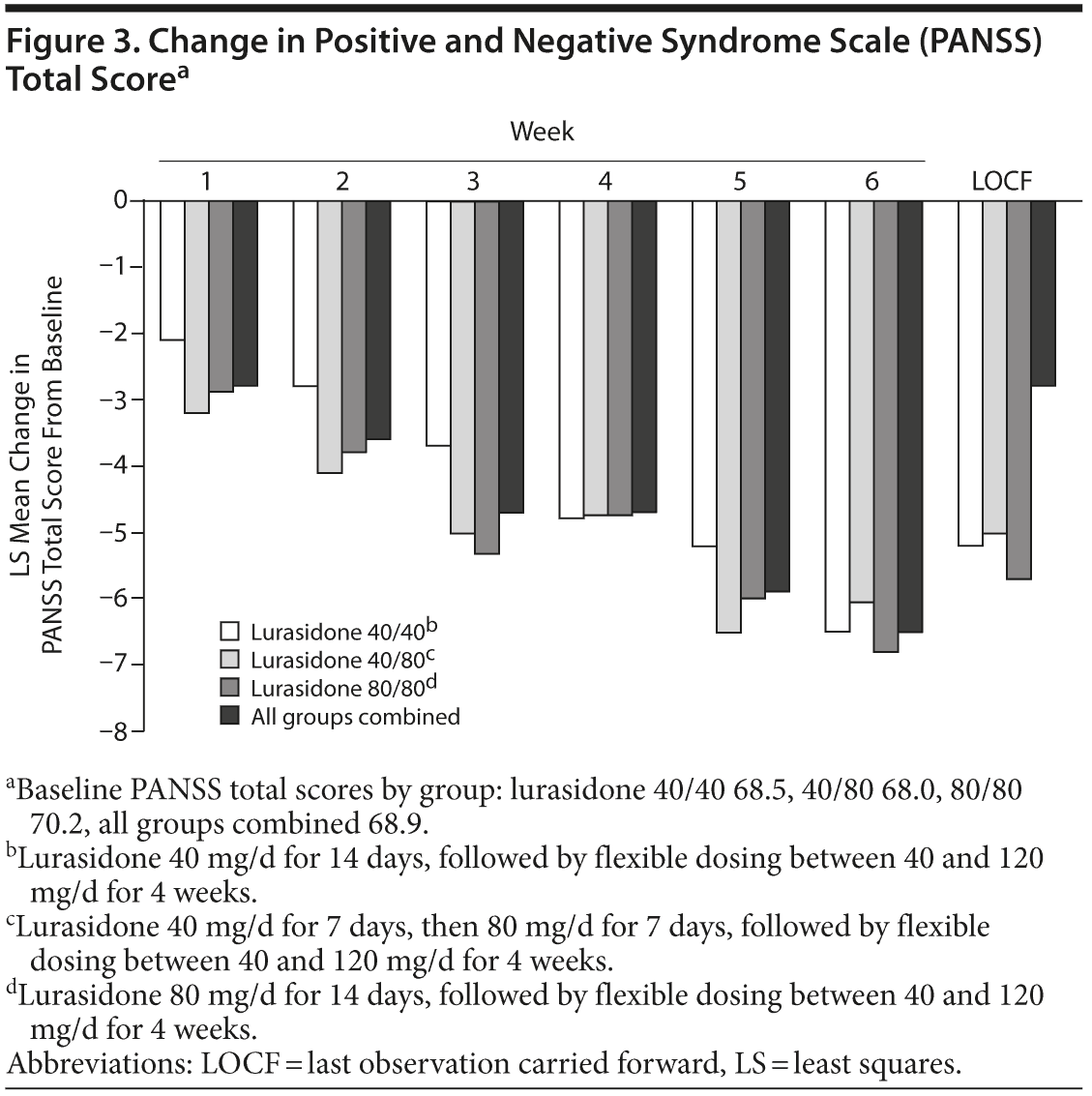
PANSS, CGI-S, and CDSS outcomes are presented in Supplementary eTable 6. The mean changes in these scores from baseline to LOCF endpoint were similar across all randomized groups. Among all subjects, mean changes in score were as follows: PANSS total, −5.8; CGI-S, −0.3; and CDSS, −1.3. Within-group improvements achieved statistical significance and demonstrated an overall effect size of 0.5 (Cohen d) on change in total score for the PANSS and 0.4 for the CGI-S and CDSS at the week 6 LOCF endpoint. Figure 3 displays the change in PANSS total score by study visit over time.
DISCUSSION
This is the first study to systematically examine the effects of switching clinically stable, but symptomatic, patients with schizophrenia or schizoaffective disorder to lurasidone. While lurasidone registration trials1–4 were limited to patients with schizophrenia, in this study approximately 37% of subjects were diagnosed with schizoaffective disorder, enhancing the generalizability of this study to patients commonly encountered in clinical practice.
In the face of suboptimal response or adverse effects, switching antipsychotic medications is often contemplated as a means of gaining improved overall effectiveness in patients with schizophrenia. Although the addition of a second medication is sometimes suggested,23 monotherapy, wherever possible, is simpler, is less costly, and can facilitate both adherence and weight loss.24,25
The primary outcome measure in this switching study was time to treatment failure (prospectively defined as any occurrence of insufficient clinical response, exacerbation of underlying disease, or discontinuation due to an AE). Time to treatment failure offers an integrated measure of efficacy and tolerability that is clinically relevant and is perhaps more meaningful than using as a primary outcome measure a rating scale that measures psychopathology.26 Time to treatment failure has been proposed to more accurately reflect drug effects, compared with all-cause discontinuation, because the latter outcome can include discontinuations that are not necessarily related to failure of the intervention. For example, it has been argued that in the context of the CATIE schizophrenia study, discontinuations “owing to patient’s decision” (a component of all-cause discontinuation) may have reflected effects that were unrelated to drug treatment, such as subject dissatisfaction with study participation.27
In this study, switching to lurasidone in outpatients treated with a broad range of antipsychotic agents was safe and effective, with low rates of study discontinuation. No meaningful differences in time to treatment failure were observed when comparing the groups of subjects randomized to start with lurasidone 40 mg/d for 2 weeks, versus starting at 80 mg/d for 2 weeks, versus starting at 40 mg/d for 1 week followed by 80 mg/d for the second week.
Improvements in psychopathologic outcomes were observed. Although baseline mean scores were low (PANSS total = 68.9, CGI-S = 3.7, CDSS total = 3.6), moderate improvement was noted for the overall sample, with effect sizes of 0.5 (Cohen d) for the PANSS and 0.4 for the CGI-S and CDSS. However, this improvement is somewhat difficult to interpret, as this open-label study did not include a parallel control group. In this context, symptomatic improvement may be attributable to the effects of time and of receiving care in a structured study environment, as well as to specific drug-related improvement.
The most commonly encountered AE was nausea (13.8% of all subjects); this was not necessarily associated with vomiting, which was reported in 7.1% of subjects. Insomnia was reported in this study in 12.9% of subjects, which is higher than the 8% observed in lurasidone schizophrenia registration trials.1 Akathisia was reported as an AE in 12.5% of all subjects, which is consistent with the rate of 13% reported in product labeling.1 One subject (0.4%) discontinued the study because of akathisia. However, median and mean BAS scores did not demonstrate meaningful changes, and, at LOCF endpoint, 205/231 (88.7%) of subjects had a BAS score rated as “absent,” 9/231 (3.9%) had a rating of “questionable,” and 14/231 (6.1%) had a rating of “mild.” A small number, 3/231 (1.3%), had a rating of “moderate,” and none of the subjects had a rating of “marked” or “severe.” Compared with baseline scores, BAS global assessment scores at LOCF endpoint were worsened for 16/231 subjects (6.9%), were improved for 24/231 (10.4%), and remained the same for 191/231 (82.7%). Overall, no clear dose-response was observed for AEs when the 3 initially randomized dose groups were compared. However, this observation was possibly confounded by the different properties of the antecedent antipsychotics; moreover, lurasidone was flexibly dosed during the last 4 weeks of the study.
Some differences emerged depending on whether the prestudy medication was classified as sedating (ie, olanzapine or quetiapine) or nonsedating (all others). The finding that insomnia rates were higher among subjects who were switched from olanzapine or quetiapine versus all other agents (18.6% vs 9.7%, respectively) appears consistent with previous reports regarding rebound insomnia after switching from sedating (ie, high affinity for H1 receptors) to nonsedating (low affinity for H1 receptors) psychotropic agents.28 Potential cholinergic rebound is also a possible concern during switches from olanzapine or quetiapine; this may have contributed to the difference in rates of vomiting seen after switch from olanzapine or quetiapine versus all other agents (9.3% vs 5.8%, respectively).28
The weight and metabolic profile of lurasidone was associated with some observed numeric improvement over the course of this 6-week study, consistent with previous findings from short and longer-term trials involving this agent.1 Lurasidone may thus be a logical antipsychotic to switch to in the presence of antipsychotic-associated weight gain. Switching to an agent with lower metabolic liability, a strategy suggested in the Patient Outcomes Research Team (PORT) recommendations,29 has previously been supported by switch studies with agents such as ziprasidone11 and aripiprazole,10,30 as well as by the Phase 2T report from the CATIE trials.31
As noted in reports of switching studies with other agents, a concern has been that switching patients from one antipsychotic to another can lead to tolerability problems, transient symptom exacerbations, or increased use of acute-care services.7,32 Su et al33 describe that adverse events can stem from the complex pharmacology in which antipsychotics target various receptor subtypes (eg, D2, 5-HT2A, M1, α1, H1) with varying degrees of affinity and that long-term antagonism of these receptors can result in physiologic counter-adaptations, such as receptor up-regulation. Thus, the 2 principal considerations when planning a switch are (1) the target dose and timing for dose escalation for the medication to switch to and (2) the timing of dose reduction and discontinuation of the medication to be switched from. Both of these key factors were explored in the current study. We did not observe any systematic efficacy or tolerability issues in the switches to lurasidone. No new safety concerns were identified.
We used the same 2-week dose reduction and discontinuation schedule for the antecedent antipsychotic in order to test any possible differences in outcome when selecting different target dosing strategies for lurasidone. This switching method differs from those used in other studies, such as a study32 of aripiprazole in which the prior antipsychotic was either immediately discontinued or tapered over 2 weeks, a study34 of olanzapine in which subjects were assigned to either abrupt discontinuation or gradual discontinuation over 2 weeks of their prior antipsychotic drug, and a series of 3 studies35 of ziprasidone in which the prior antipsychotic was completely discontinued prior to initiation of ziprasidone, the prior antipsychotic was cross-titrated with ziprasidone, or the dose reduction was delayed. Since the precise switching strategy across these prior switch studies did not consistently impact outcome,32,34,35 we believe our choice of keeping constant the method of discontinuing the antecedent antipsychotic medication to be justified. However, as a caveat, for the individual patient, the method of discontinuation from the prior antipsychotic may have clinical importance. For example, Weiden36 and Lambert37 suggest that tapering the prior medication while simultaneously increasing the dose of the newer treatment is preferable when stable outpatients are experiencing significant and troublesome side effects from their existing medication. However, an alternate method of establishing the patient on a therapeutic dose of the new medication before reducing the prior medication avoids exposure to subtherapeutic dosages and may be the safest switching method in cases in which relapse is a concern.36,37 In the individual care of patients, the optimal length of time for a cross-titration is highly dependent on the patient’s clinical status, preferences, and prior history; for some patients, the process may take only a few days, but for others, it may need to occur over a period substantially longer than the 1 to 2 weeks that switch studies pragmatically allot for in their study designs.
Additional information regarding switching strategies between second-generation antipsychotics, including withdrawal syndromes and pharmacokinetic considerations, as well as specific information about switching due to relapse; limited efficacy of the previous antipsychotic; tolerability issues such as extrapyramidal symptoms, tardive dyskinesia, weight gain, metabolic disorders, hyperprolactinemia, sexual dysfunction; or therapeutic noncompliance, can be found in a review by Edlinger et al38 and in a recent position statement authored by an expert group from psychiatric professional societies in Spain.39 Guidelines from the British Association for Psychopharmacology,40 World Federation of Societies of Biological Psychiatry,41,42 and American Psychiatric Association43 also offer switching advice. Specific guidance for switching strategies is also available through an online tool, SwitchRx (http://switchrx.ca/).
The current US product label1 for lurasidone provides for a dosing range of 40–160 mg/d. It would be reasonable to expect that switching outpatients to initial lurasidone doses of 40 or 80 mg/d would be appropriate. In our study, after week 2 was completed, the investigators were free to dose flexibly within the range of 40–120 mg/d according to the perceived tolerability and efficacy of lurasidone experienced by the individual subject. Although the most commonly used dose of lurasidone in this study was 80 mg/d, 29.2% of participants received a modal dose of 120 mg/d over the course of the trial.
Several study limitations should be noted. This study was of 6 weeks’ duration, which may not have been long enough to capture the full range of postswitch changes in safety and efficacy parameters. A 6-month extension to this study will be separately reported. Group differences in time to treatment failure may have been obscured by investigator awareness that all subjects were treated with open-label lurasidone. It is possible that individual subjects would have benefited from a more extended cross-titration period (ie, longer than 2 weeks), which was not feasible in the context of a
short-term study. It is difficult to interpret observed improvement in treatment effectiveness in the absence of a parallel control group.
The categorization of olanzapine and quetiapine as “sedating” and all other previously administered antipsychotics as “nonsedating,” although based on what is known about these agents and their relative propensity to be associated with sedation and/or somnolence,28,44 does not take into account all potential differences within and between these 2 broad groups. It is arguable that chlorpromazine should also have been included in the sedating group; however, only 3 subjects were receiving chlorpromazine as their primary antipsychotic at the time of enrollment. The lack of available information on preswitch sedation status is also a limiting factor when considering the validity of the sedating versus nonsedating distinction regarding preswitch agents made in this study. However, observed differences in outcome after the switch to lurasidone from agents classified in this study on the basis of their sedating versus nonsedating properties suggest that this distinction may be clinically relevant. Stratification on the basis of single antipsychotics and/or pharmacologic properties other than sedation may yield different results than reported here.
CONCLUSIONS
Switching patients to lurasidone can be successfully accomplished by starting at 40 mg/d for 2 weeks, or 80 mg/d for 2 weeks, or 40 mg/d for 1 week followed by 80 mg/d the second week. Over 80% of subjects remained in the study at the end of the 6 weeks. Treatment failures constituted less than 8% of the study population. Among the 3 lurasidone dosing strategies, there were no clinically meaningful differences in treatment failure, all-cause discontinuation, AEs, or metabolic variables or differences on rating scales including the BAS, SAS, AIMS, PANSS, CGI-S, and CDSS. Improvement in weight and lipid variables was observed after switch to lurasidone in this short-term study. The properties of the antecedent antipsychotic may influence the incidence of AEs with lurasidone. Switching should thus be tailored to the specific individual and attention should be paid to the emergence of insomnia in persons who had been receiving a sedating antipsychotic immediately prior to lurasidone.
Drug names: aripiprazole (Abilify), asenapine (Saphris), benztropine (Cogentin and others), biperiden (Akineton), diphenhydramine (Benadryl and others), divalproex (Depakote and others), eszopiclone (Lunesta), haloperidol (Haldol and others), iloperidone (Fanapt), lamotrigine (Lamictal and others), lithium (Lithobid and others), lorazepam (Ativan and others), lurasidone (Latuda), olanzapine (Zyprexa), paliperidone (Invega), propranolol (Inderal, InnoPran, and others), quetiapine (Seroquel), risperidone (Risperdal and others), temazepam (Restoril and others), thiothixene (Navane and others), zaleplon (Sonata and others), ziprasidone (Geodon), zolpidem (Ambien, Edluar, and others).
Author affiliations: Department of Psychiatry and Behavioral Sciences, Duke University Medical Center, Durham, North Carolina (Dr McEvoy); Department of Psychiatry and Behavioral Sciences, New York Medical College, Valhalla, New York (Dr Citrome); and Sunovion Pharmaceuticals Inc, Fort Lee, New Jersey (Mr Hernandez and Drs Cucchiaro, Hsu, Pikalov, and Loebel).
Principal investigators at study sites: Prakash Bhatia, MD, Synergy Clinical Research of Escondido, Escondido, CA; Brian Bortnick, MD, Comprehensive Neuroscience Inc, Atlanta, GA; Ronald Brenner, MD, Neurobehavioral Research, Inc, Cedarhurst, NY; Peter Buckley, MD, Georgia Health Sciences University, Augusta, GA; Marina Bussel, MD, California Clinical Trials, Paramount, CA; Matthew Byerly, MD, University of Texas Southwestern Medical Center, Dallas, TX; John Canale, MD, Saint Charles Psychiatric Associates/Midwest Research Group, St Charles, MO; Leslie Citrome, MD, MPH, Nathan Kline Institute, Orangeburg, NY; Michael Downing, MD, FutureSearch Trials of Dallas, Dallas, TX; David Feifel, MD, PhD, University of California San Diego Medical Center, San Diego, CA; Carlos Figueroa, MD, Pasadena Research Institute, Pasadena, CA; Donald Garcia Jr, MD, FutureSearch Clinical Trials, LP, Austin, TX; Ira Glick, MD Pacific Research Partners, LLC, Oakland, CA; Armen Goenjian, MD, Collaborative Neuroscience Network, South Bay, Torrance, CA; Daniel Gruener, MD, CRI Worldwide-Kirkbride Center, Philadelphia, PA; Thomas Grugle, MD, Pillar Clinical Research, LLC, Dallas, TX; Greg Kaczenski, K and S Professional Research Services, Little Rock, AR; Raymond Manning, MD, California Neuropsychopharmacology Clinical Research Institute (CNRI), LLC, Pico Rivera, CA; James Kimball, MD, Wake Forest University Baptist Medical Center, Winston-Salem, NC; Joseph McEvoy, MD, Duke University Medical Center, Durham, NC; Nilesh Patel, MD, Wharton Research Center, Wharton, TX; Georgios Petrides, MD, Zucker Hillside Hospital—NorthShore-LIJ Heath System, Glen Oaks, NY; Fred Reimherr, MD, University of Utah School of Medicine, Salt Lake City, UT; Tram Tran-Johnson, PharmD, PsyD, California Neuropsychopharmacology Clinical Research Institute (CNRI), San Diego, CA; David Walling, PhD, Collaborative Neuroscience Network, Inc, Garden Grove, CA; Peter Weiden, MD, University of Illinois at Chicago, Chicago, IL; Brian Wise, MD, MPH, Western Affiliated Research Institute, Denver, CO; and Kashinath Yadalam, MD, Lake Charles Clinical Trials, LLC,
Lake Charles, LA.
Potential conflicts of interest: In the past 12 months, Dr McEvoy has been a consultant for, received honoraria from, or conducted clinical research supported by Alkermes, Eli Lilly, Merck, Psychogenics, Roche, and Sunovion. In the past 12 months, Dr Citrome has been a consultant for, received honoraria from, or conducted clinical research supported by Alexza, Alkermes, AstraZeneca, Avanir, Bristol-Myers Squibb, Eli Lilly, Forest, Genentech, Janssen, Lundbeck, Merck, Novartis, Noven, Otsuka, Pfizer, Shire, and Sunovion and holds small numbers of shares in Bristol-Myers Squibb, Eli Lilly, Johnson & Johnson, Merck, and Pfizer. Mr Hernandez and Drs Cucchiaro, Hsu, Pikalov, and Loebel are full-time employees of Sunovion.
Funding/support: This study was supported by Sunovion Pharmaceuticals Inc, Marlborough, Massachusetts, and Fort Lee, New Jersey.
Acknowledgments: The authors thank the patients who participated
in this study, as well as the principal investigators at the study sites.
Supplementary material: Available at PSYCHIATRIST.COM.
REFERENCES
1. Sunovion. Latuda (lurasidone). http://www.latuda.com/LatudaPrescribingInformation.pdf. Revised April 2012. Updated May 2012. Accessed December 13, 2012.
2. Nakamura M, Ogasa M, Guarino J, et al. Lurasidone in the treatment of acute schizophrenia: a double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(6):829–836. doi:10.4088/JCP.08m04905 PubMed
3. Meltzer HY, Cucchiaro J, Silva R, et al. Lurasidone in the treatment of schizophrenia: a randomized, double-blind, placebo- and olanzapine-controlled study. Am J Psychiatry. 2011;168(9):957–967. doi:10.1176/appi.ajp.2011.10060907 PubMed
4. Ogasa M, Kimura T, Nakamura M, et al. Lurasidone in the treatment of schizophrenia: a 6-week, placebo-controlled study. Psychopharmacology (Berl). 2012. doi:10.1007/s00213-012-2838-2 PubMed
5. Citrome L. Lurasidone for the acute treatment of adults with schizophrenia: what is the number needed to treat, number needed to harm, and likelihood to be helped or harmed? Clin Schizophr Relat Psychoses. 2012;6(2):76–85. doi:10.3371/CSRP.6.2.5 PubMed
6. Volavka J, Citrome L. Oral antipsychotics for the treatment of schizophrenia: heterogeneity in efficacy and tolerability should drive decision-making. Expert Opin Pharmacother. 2009;10(12):1917–1928. doi:10.1517/14656560903061309 PubMed
7. Faries DE, Ascher-Svanum H, Nyhuis AW, et al. Clinical and economic ramifications of switching antipsychotics in the treatment of schizophrenia. BMC Psychiatry. 2009;9(1):54. doi:10.1186/1471-244X-9-54 PubMed
8. Covell NH, Jackson CT, Evans AC, et al. Antipsychotic prescribing practices in Connecticut’s public mental health system: rates of changing medications and prescribing styles. Schizophr Bull. 2002;28(1):17–29. doi:10.1093/oxfordjournals.schbul.a006920 PubMed
9. Citrome L. Interpreting and applying the CATIE results: with CATIE, context is key, when sorting out Phases 1, 1A, 1B, 2E, and 2T. Psychiatry (Edgmont). 2007;4(10):23–29. PubMed
10. Stroup TS, McEvoy JP, Ring KD, et al; Schizophrenia Trials Network. A randomized trial examining the effectiveness of switching from olanzapine, quetiapine, or risperidone to aripiprazole to reduce metabolic risk: Comparison Of Antipsychotics for Metabolic Problems (CAMP).
Am J Psychiatry. 2011;168(9):947–956. doi:10.1176/appi.ajp.2011.10111609 PubMed
11. Weiden PJ, Newcomer JW, Loebel AD, et al. Long-term changes in
weight and plasma lipids during maintenance treatment with ziprasidone. Neuropsychopharmacology. 2008;33(5):985–994. doi:10.1038/sj.npp.1301482 PubMed
12. Guy W. ECDEU Assessment Manual for Psychopharmacology, Revised. US Department of Health, Education and Welfare publication (ADM) 76-338. Rockville, MD: National Institute of Mental Health; 1976:534–537.
13. Munetz MR, Benjamin S. How to examine patients using the Abnormal Involuntary Movement Scale. Hosp Community Psychiatry. 1988;39(11):
1172–1177. PubMed
14. Barnes TRE. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154(5):672–676. doi:10.1192/bjp.154.5.672 PubMed
15. Simpson GM, Angus JW. A rating scale for extrapyramidal side effects.
Acta Psychiatr Scand suppl. 1970;212(S212):11–19. doi:10.1111/j.1600-0447.1970.tb02066.x PubMed
16. Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale Manual. North Tonawanda, NY: Multi-Health Systems; 1994.
17. Addington D, Addington J, Schissel B. A depression rating scale for schizophrenics. Schizophr Res. 1990;3(4):247–251. doi:10.1016/0920-9964(90)90005-R PubMed
18. Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):
1266–1277. doi:10.1176/appi.ajp.2011.10111704 PubMed
19. Voruganti LN, Awad AG. Personal Evaluation of Transitions in Treatment (PETiT): a scale to measure subjective aspects of antipsychotic drug therapy in schizophrenia. Schizophr Res. 2002;56(1-2):37–46. doi:10.1016/S0920-9964(01)00161-X PubMed
20. Browne G, Gafni A, Roberts J, et al. Approach to the Measurement of
Costs (Expenditures) When Evaluating the Efficiency of Health and Social Programmes. Working Paper Series 92-12, System-Linked Research Unit. Hamilton, Ontario, Canada: McMaster University; 1992.
21. Ware J Jr, Kosinski M, Keller SD. A 12-Item Short-Form Health Survey: construction of scales and preliminary tests of reliability and validity.
Med Care. 1996;34(3):220–233. doi:10.1097/00005650-199603000-00003 PubMed
22. Vernon MK, Revicki DA, Awad AG, et al. Psychometric evaluation of the Medication Satisfaction Questionnaire (MSQ) to assess satisfaction with antipsychotic medication among schizophrenia patients. Schizophr Res. 2010;118(1–3):271–278. doi:10.1016/j.schres.2010.01.021 PubMed
23. Citrome L. Treatment-refractory schizophrenia: what is it and
what has been done about it? Neuropsychiatry. 2011;1(4):325–347. doi:10.2217/npy.11.35
24. Burton SC. Strategies for improving adherence to second-generation antipsychotics in patients with schizophrenia by increasing ease of use. J Psychiatr Pract. 2005;11(6):369–378. doi:10.1097/00131746-200511000-00003 PubMed
25. Essock SM, Schooler NR, Stroup TS, et al; Schizophrenia Trials Network. Effectiveness of switching from antipsychotic polypharmacy to monotherapy. Am J Psychiatry. 2011;168(7):702–708. doi:10.1176/appi.ajp.2011.10060908 PubMed
26. Glick ID, Correll CU, Altamura AC, et al. Mid-term and long-term
efficacy and effectiveness of antipsychotic medications for schizophrenia:
a data-driven, personalized clinical approach. J Clin Psychiatry. 2011;72(12):
1616–1627. doi:10.4088/JCP.11r06927 PubMed
27. Kraemer HC, Glick ID, Klein DF. Clinical trials design lessons from the CATIE study. Am J Psychiatry. 2009;166(11):1222–1228. doi:10.1176/appi.ajp.2009.08121809 PubMed
28. Buckley PF. Receptor-binding profiles of antipsychotics: clinical strategies when switching between agents. J Clin Psychiatry. 2007;68(suppl 6):5–9. PubMed
29. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71–93. doi:10.1093/schbul/sbp116 PubMed
30. Newcomer JW, Campos JA, Marcus RN, et al. A multicenter, randomized, double-blind study of the effects of aripiprazole in overweight subjects
with schizophrenia or schizoaffective disorder switched from olanzapine. J Clin Psychiatry. 2008;69(7):1046–1056. doi:10.4088/JCP.v69n0702 PubMed
31. Stroup TS, Lieberman JA, McEvoy JP, et al; CATIE Investigators. Effectiveness of olanzapine, quetiapine, risperidone, and ziprasidone in patients with chronic schizophrenia following discontinuation of a previous atypical antipsychotic. Am J Psychiatry. 2006;163(4):611–622. doi:10.1176/appi.ajp.163.4.611 PubMed
32. Casey DE, Carson WH, Saha AR, et al; Aripiprazole Study Group. Switching patients to aripiprazole from other antipsychotic agents: a multicenter randomized study. Psychopharmacology (Berl). 2003;166(4):391–399. PubMed
33. Su J, Barr AM, Procyshyn RM. Adverse events associated with switching antipsychotics. J Psychiatry Neurosci. 2012;37(1):E1–E2. doi:10.1503/jpn.110096 PubMed
34. Kinon BJ, Basson BR, Gilmore JA, et al. Strategies for switching
from conventional antipsychotic drugs or risperidone to olanzapine.
J Clin Psychiatry. 2000;61(11):833–840. doi:10.4088/JCP.v61n1105 PubMed
35. Weiden PJ, Simpson GM, Potkin SG, et al. Effectiveness of switching
to ziprasidone for stable but symptomatic outpatients with schizophrenia.
J Clin Psychiatry. 2003;64(5):580–588. doi:10.4088/JCP.v64n0514 PubMed
36. Weiden PJ. Switching antipsychotics: an updated review with a focus
on quetiapine. J Psychopharmacol. 2006;20(1):104–118. doi:10.1177/0269881105056668 PubMed
37. Lambert TJ. Switching antipsychotic therapy: what to expect and clinical strategies for improving therapeutic outcomes. J Clin Psychiatry. 2007;
68(suppl 6):10–13. PubMed
38. Edlinger M, Baumgartner S, Eltanaihi-Furtmüller N, et al. Switching between second-generation antipsychotics: why and how? CNS Drugs. 2005;19(1):
27–42. doi:10.2165/00023210-200519010-00003 PubMed
39. Bernardo M, Vieta E, Ruiz JS, et al. Recommendations for switching antipsychotics. A position statement of the Spanish Society of Psychiatry and the Spanish Society of Biological Psychiatry. Rev Psiquiatr Salud Ment (Barc). 2011;4:150–168. doi:10.1016/j.rpsm.2011.07.003
40. Barnes TR; Schizophrenia Consensus Group of British Association for Psychopharmacology. Evidence-based guidelines for the pharmacological treatment of schizophrenia: recommendations from the British Association for Psychopharmacology. J Psychopharmacol. 2011;25(5):567–620. doi:10.1177/0269881110391123 PubMed
41. Falkai P, Wobrock T, Lieberman J, et al; WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia,
pt 2: long-term treatment of schizophrenia. World J Biol Psychiatry. 2006;
7(1):5–40. doi:10.1080/15622970500483177 PubMed
42. Falkai P, Wobrock T, Lieberman J, et al; WFSBP Task Force on Treatment Guidelines for Schizophrenia. World Federation of Societies of Biological Psychiatry (WFSBP) guidelines for biological treatment of schizophrenia,
pt 1: acute treatment of schizophrenia. World J Biol Psychiatry. 2005;6(3):
132–191. doi:10.1080/15622970510030090 PubMed
43. Lehman AF, Lieberman JA, Dixon LB, et al; American Psychiatric Association; Steering Committee on Practice Guidelines. Practice
Guideline for the Treatment of Patients With Schizophrenia, Second Edition.
Am J Psychiatry. 2004;161(2 suppl):1–56. PubMed
44. Citrome L, Nasrallah HA. On-label on the table: what the package insert informs us about the tolerability profile of oral atypical antipsychotics, and what it does not. Expert Opin Pharmacother. 2012;13(11):1599–1613. doi:10.1517/14656566.2011.626767 PubMed
![]() Save
Save
![]() Share
Share
![]() Cite
Cite
Advertisement
GAM ID: sidebar-top