This work may not be copied, distributed, displayed, published, reproduced, transmitted, modified, posted, sold, licensed, or used for commercial purposes. By downloading this file, you are agreeing to the publisher’s Terms & Conditions.
Article Abstract
Objective: Depression is the predominant psychosocial and suicide burden in bipolar disorder, yet there is a paucity of evidence-based treatments for bipolar depression.
Methods: This post hoc subgroup analysis of data pooled from two 3-week, randomized, placebo- and olanzapine-controlled trials (December 2004-April 2006, N = 489 and November 2004-April 2006, N = 488) examined a subgroup of patients meeting criteria for moderate-to-severe mixed major depressive episodes, defined using DSM-IV-TR criteria for mixed episodes (mania and major depression simultaneously) with a baseline Montgomery-Asberg Depression Rating Scale (MADRS) total score ≥ 20.
Results: Decreases in MADRS scores (least squares mean [SE]), the a priori primary outcome, were significantly greater in the asenapine group than in the placebo group from baseline to day 7 (-11.02 [1.82] vs -4.78 [1.89]; P = .0195), day 21 (-14.03 [2.01] vs -7.43 [2.09]; P = .0264), and endpoint (-10.71 [1.76] vs -5.19 [1.98]; P = .039). Decreases in MADRS scores with asenapine were significantly greater than with olanzapine from baseline to day 7 (-6.26 [1.47]; P = .0436). Decreases in Young Mania Rating Scale mean total score were greater with asenapine than with placebo or olanzapine at all time points assessed. A significantly greater reduction from baseline to day 21 in the Short Form-36 mental component summary score was observed with asenapine, but not olanzapine, compared with placebo (16.57 vs 5.97; P = .0093). Asenapine was generally well tolerated.
Conclusions: These data provide support for the potential efficacy of asenapine in mixed major depressive episodes; however, these data cannot be linearly extrapolated to nonmixed major depression.
ABSTRACT
Objective: Depression is the predominant psychosocial and suicide burden in bipolar disorder, yet there is a paucity of evidence-based treatments for bipolar depression.
Methods: This post hoc subgroup analysis of data pooled from two 3-week, randomized, placebo- and olanzapine-controlled trials (December 2004-April 2006, N = 489 and November 2004-April 2006, N = 488) examined a subgroup of patients meeting criteria for moderate-to-severe mixed major depressive episodes, defined using DSM-IV-TR criteria for mixed episodes (mania and major depression simultaneously) with a baseline Montgomery-Asberg Depression Rating Scale (MADRS) total score ≥ 20.
Results: Decreases in MADRS scores (least squares mean [SE]), the a priori primary outcome, were significantly greater in the asenapine group than in the placebo group from baseline to day 7 (-11.02 [1.82] vs -4.78 [1.89]; P = .0195), day 21 (-14.03 [2.01] vs -7.43 [2.09]; P = .0264), and endpoint (-10.71 [1.76] vs -5.19 [1.98]; P = .039). Decreases in MADRS scores with asenapine were significantly greater than with olanzapine from baseline to day 7 (-6.26 [1.47]; P = .0436). Decreases in Young Mania Rating Scale mean total score were greater with asenapine than with placebo or olanzapine at all time points assessed. A significantly greater reduction from baseline to day 21 in the Short Form-36 mental component summary score was observed with asenapine, but not olanzapine, compared with placebo (16.57 vs 5.97; P = .0093). Asenapine was generally well tolerated.
Conclusions: These data provide support for the potential efficacy of asenapine in mixed major depressive episodes; however, these data cannot be linearly extrapolated to nonmixed major depression.
J Clin Psychiatry 2015;76(6):728-734
© Copyright 2015 Physicians Postgraduate Press, Inc.
Submitted: October 4, 2013; accepted May 16, 2014.
Online ahead of print: January 20, 2015 (doi:10.4088/JCP.13m08827).
Corresponding author: Michael Berk, MD, PhD, School of Medicine, Deakin University, 1 Gheringhap St, Geelong, VIC 3220, Australia ([email protected]).
Bipolar I disorder is characterized by mania and/or mixed episodes, but patients predominantly experience depressive episodes.1 Studies over the past 2 decades that have prospectively monitored the symptomatic status of patients with bipolar I disorder suggest that depressive symptoms are 3 times more common than manic/hypomanic symptoms and that syndromal/subsyndromal depressive symptoms are present at least 50% of the time.2,3 Consequently, the depressive symptoms are particularly disabling, as they diminish the quality of life and result in marked social and occupational impairment.4 Notably, in addition to depression, depressive mixed states confer the greatest suicide risk.5-7 These risks and the debilitating sequelae of bipolar depression stem from heterogeneous etiologic factors, which, in turn, are the product of underlying biological vulnerability interacting with developmental factors, personality, and cognitive factors, which together generate a stress diathesis sensitive to lifestyle factors and substance misuse.8-14 Clearly, these etiologic variances and responses to prior treatments need to be taken into account when developing personalized treatment packages.15
Remarkably, despite the predominance of depression, few proven treatments exist for the management of depressive symptoms in this population.16 Although antidepressant medications are widely used in clinical practice in combination with mood stabilizers or atypical antipsychotics to treat depressive symptoms in bipolar patients, only 3 large double-blind trials have examined their efficacy.17-19 In 2 of these trials, adjunctive antidepressants were not superior to placebo adjunctive therapy with mood stabilizers.17,18 Hence, only 2 treatments (quetiapine monotherapy and olanzapine-plus-fluoxetine combination) are approved by the US Food and Drug Administration for bipolar depression. Other atypical antipsychotics, such as aripiprazole and ziprasidone, failed to separate from placebo in clinical trials.20,21 Recently, lurasidone monotherapy and lurasidone adjunctive to lithium or valproate significantly reduced depressive symptoms in patients with bipolar I depression.22,23 Still, the treatment of bipolar depression remains a critical unmet need.24,25
The aim of this analysis was to examine post hoc the efficacy of asenapine, a novel atypical antipsychotic, in the treatment of depressive symptoms in the subgroup of individuals meeting criteria for a moderate-to-severe major depressive episode concurrent with an episode of mania (mixed major depressive episode).
METHOD
The data for this post hoc subgroup analysis were obtained from the two 3-week, randomized, double-blind, multicenter, placebo- and olanzapine-controlled trials (ARES 7501005, NCT00159796, N = 489; and ARES 7501004, NCT00159744, N = 488) that examined the efficacy of asenapine in treating manic/mixed episodes in patients with bipolar I disorder.26,27 The original trials were conducted in compliance with the Declaration of Helsinki and the principles of Good Clinical Practice and were approved by the appropriate institutional review boards. All subjects enrolled in those trials provided written informed consent.26,27

- Atypical antipsychotics have potential value in bipolar depression; however, data on asenapine are lacking.
- In a post hoc analysis of patients meeting criteria for major depression while in a mixed state, there was a suggestion of utility of asenapine.
- Caution is necessary in interpreting data from post hoc analyses of a study that was not designed for that outcome.
Study Design and Patient Population
A total of 977 patients were enrolled in the 2 published studies.26,27 Patients in these studies were ≥ 18 years of age with a current Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) primary diagnosis of bipolar I disorder. Enrolled patients were experiencing manic or mixed episodes that began less than 3 months prior to screening. Their Young Mania Rating Scale (YMRS)28 total score was ≥ 20 at screening and baseline. In addition, the Mini-International Neuropsychiatric Interview29 was used to assess manic or mixed episodes. Enrolled patients had a documented history of ≥ 1 moderate-to-severe mood episode with or without psychotic features. Key exclusion criteria included a psychiatric diagnosis or primary diagnosis other than bipolar disorder, rapid-cycling bipolar disorder in the year before screening, and a current DSM-IV-TR diagnosis of substance abuse or dependence.
Each of the 2 studies consisted of a 1-week run-in/washout period that preceded enrollment, after which eligible subjects were randomized to asenapine:placebo:olanzapine in a 2:1:2 ratio. Additional psychotropic medications were not permitted during the trials, except for benzodiazepines and non-benzodiazepine sedative-hypnotics, which were allowed during the run-in period and the first week of treatment.
Treatments
Treatments were administered daily for 3 weeks in a double-dummy manner, using 3 film-coated tablets (for olanzapine or placebo) and 2 sublingual fast-dissolving tablets (for asenapine or placebo). Subjects received asenapine 20 mg daily on day 1 and then flexible 10 or 20 mg daily thereafter, divided into 1 morning and 1 evening dose. Olanzapine treatment consisted of 15 mg once daily on day 1 and then flexible 5 to 20 mg daily thereafter.
Assessments
The primary efficacy outcome of the 2 studies was change from baseline to day 21 in YMRS total score compared with placebo, which was met in both studies. The change from baseline in ARES 7501005 was −10.8 vs −5.5 for placebo (P ≤ .0001), and the change from baseline in ARES 7501004 was −11.5 vs −7.8 for placebo (P < .007). Secondary efficacy outcomes were change from baseline in Clinical Global Impression for Bipolar Disorder scale score, change from baseline in depressive symptoms using Montgomery-Asberg Depression Rating Scale (MADRS) scores,30 the percentage of YMRS responders (patients with ≥ 50% reduction from baseline in YMRS total score), and the percentage of YMRS remitters (patients with YMRS total score ≤ 12).26,27
Data on adverse events, including serious adverse events, were collected continuously throughout the studies up to 7 days after last dose intake for adverse events and up to 30 days after last dose intake for serious adverse events.
Subgroup and Outcome Measures for Post Hoc Subgroup Analysis
Patients meeting criteria for moderate-to-severe mixed major depressive episodes, defined using DSM-IV-TR criteria for mixed episode and a baseline MADRS total score ≥ 20 (of a possible maximum score of 60),31 were included in this post hoc subgroup analysis. The primary a priori outcome (defined for the purpose of this post hoc subgroup analysis) was improvement in depressive symptoms, which was evaluated as change from baseline in MADRS total and individual scores on day 7 and day 21 for each group. Secondary outcome measures included improvement in manic symptoms and quality of life. Treatment effects on manic symptoms were evaluated as change from baseline in YMRS total scores and were assessed on days 2, 4, 7, 14, and 21 or at endpoint (last observation carried forward ([LOCF]). Change from baseline in Short-Form 36 (SF-36) physical and mental summary and individual subscale scores (physical functioning, role-physical, bodily pain, and general health for physical; vitality, social functioning, role-emotional, and mental health for mental) were assessed on day 21.
Statistical Analysis
We aimed a priori to use an analytic plan that replicated as closely as possible previous strategies for similar analyses.32 Statistical methods used for subgroup analysis were the same as those used for analysis in the primary studies.26,27 For statistical analyses, comparisons were made between the asenapine group and the olanzapine and placebo groups and between the olanzapine group and placebo group. It should be noted that the studies were not powered to draw conclusions for this post hoc subgroup analysis. Analysis of efficacy parameters included all subjects who took ≥ 1 dose of study medication and had ≥ 1 postbaseline MADRS assessment. Changes from baseline in MADRS, YMRS, and SF-36 scores were expressed as least squares (LS) means. Changes from baseline in YMRS total score, MADRS total and individual item scores, and SF-36 subscale scores were analyzed using analysis of covariance models, with treatment and protocol as factors and baseline value as a covariate. Observed cases at each visit were analyzed, while LOCF was used for missing data at endpoint. Comparisons were not adjusted for multiplicity, and a 2-sided 5% level of significance was used to assess statistical significance. It should be noted that unplanned subgroup analyses tend to have a higher but unknown type I error (false positive) rate. Safety parameters, including adverse events, were assessed for all subjects receiving ≥ 1 dose of study medication and who had ≥ 1 post-baseline YMRS assessment. Treatment-emergent adverse events were coded using the MedDRA dictionary. Adverse event tabulations included the number and percentage of adverse events that occurred in ≥ 5% of subjects in the asenapine or olanzapine group and were more than twice as frequent as in the placebo group, by MedDRA preferred term. Statistical analyses were performed using SAS version 9.2 (SAS Institute, Inc, Cary, North Carolina).
RESULTS
Disposition and Demographics
Of the 977 subjects randomized in the 2 trials, 98 subjects met the inclusion criteria for this post hoc subgroup analysis (ie, a major depressive episode and a manic episode and a baseline MADRS total score ≥ 20). In this group of 98 subjects with moderate-to-severe mixed major depressive episodes, 33 received asenapine (flexible 10-mg or 20-mg total daily dose), 26 received placebo, and 39 received olanzapine (flexible 5-20 mg total daily dose). A total of 31 subjects discontinued prior to study completion, 14 in the asenapine group (4 due to adverse events, 2 due to lack of efficacy, 1 lost to follow-up, and 7 due to withdrawn consent), 7 in the placebo group (4 due to lack of efficacy and 3 due to withdrawn consent), and 10 in the olanzapine group (1 due to adverse event, 6 due to lack of efficacy, 1 lost to follow-up, and 2 due to withdrawn consent). Subject demographic information is presented in Table 1. Approximately half of the subjects in this post hoc subgroup analysis were female. The mean and median ages were numerically similar in the 3 treatment groups.
Click figure to enlarge
Effect on Depressive Symptoms (MADRS)
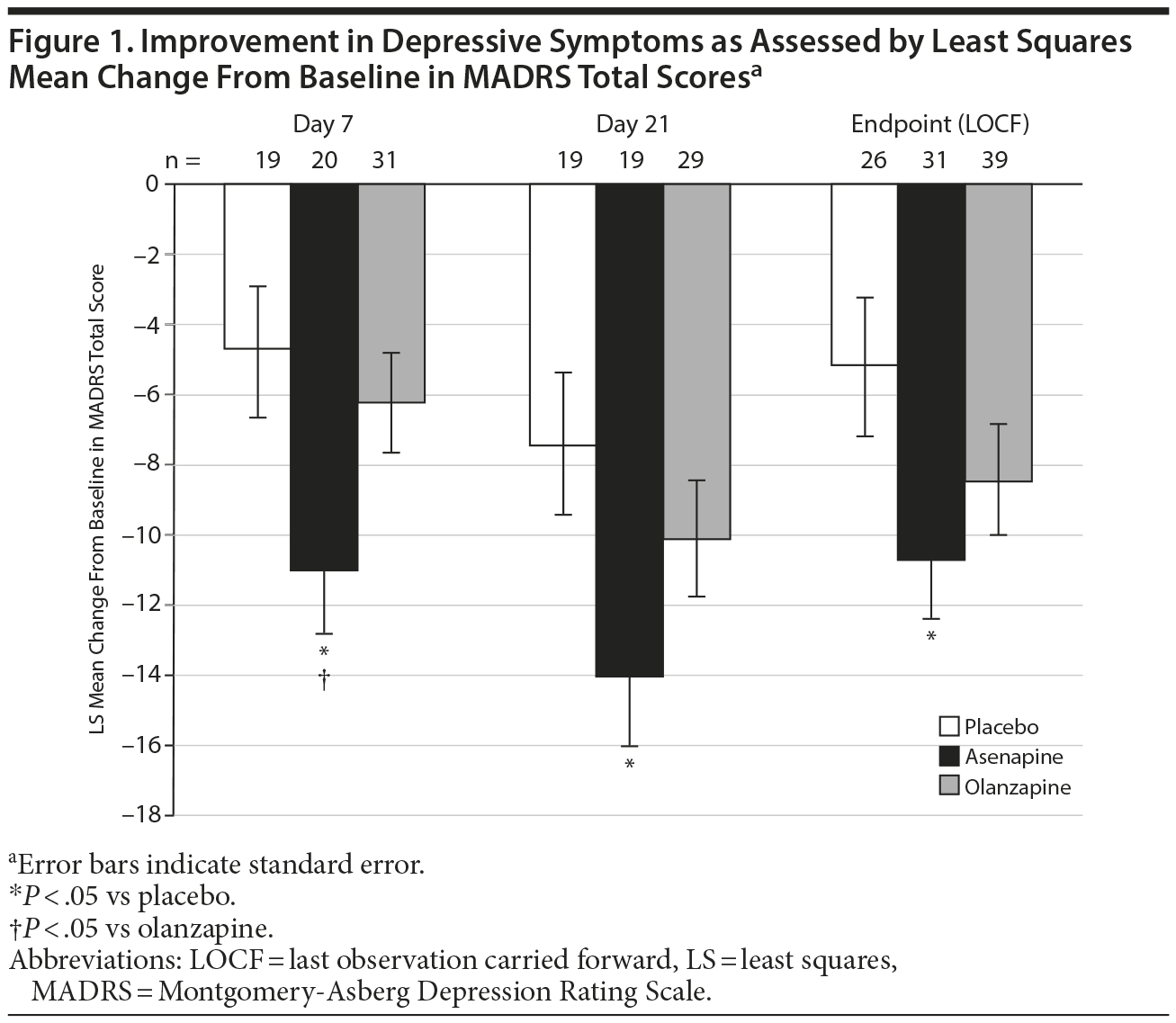
Improvement in depressive symptoms, evaluated as mean change (decrease) from baseline in MADRS total scores, was greater in the asenapine group compared with the placebo and olanzapine groups on days 7 and 21 and at the endpoint (Figure 1). Specifically, decreases in MADRS scores (LS mean [SE]) in the asenapine group were significantly greater compared with the placebo group from baseline to day 7 (-11.02 [1.82] vs -4.78 [1.89]; P = .0195; LOCF −11.16 [1.60] vs −4.85 [1.82]; P = .0103), day 21 (-14.03 [2.01] vs -7.43 [2.09]; P = .0264), and endpoint (LOCF -10.71 [1.76] vs -5.19 [1.98]; P = .039). Decreases in MADRS scores in the asenapine group were significantly greater compared with the olanzapine group from baseline to day 7 (-6.26 [1.47]; P = .0436; LOCF −5.91 [1.37]; P = .0136). However, decreases in MADRS scores in the asenapine group were not significantly different compared with the olanzapine group from baseline to day 21 (-10.12 [1.69]; P = .1333) and to the endpoint (LOCF -8.45 [1.58]; P = .3387).
Click figure to enlarge
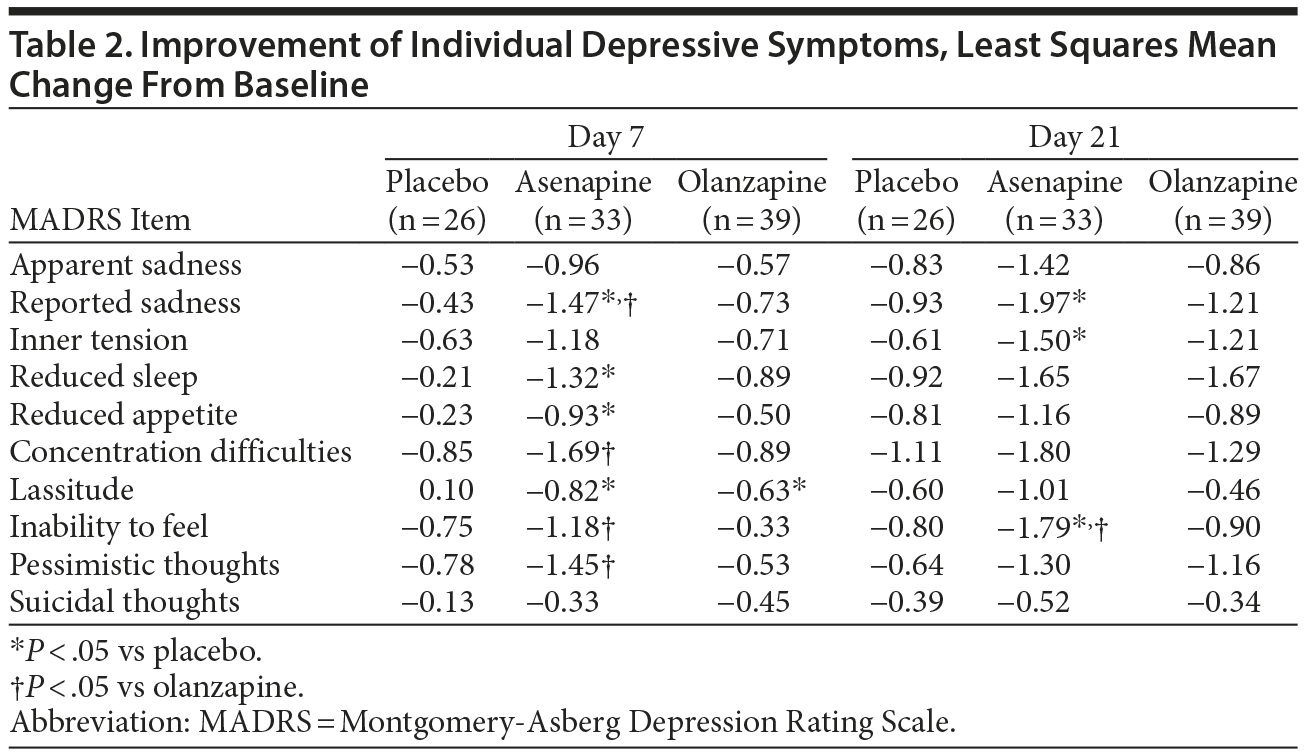
Improvements in individual depressive symptoms are presented in Table 2. By day 7, decreases were significantly greater in the asenapine group compared with the placebo group for reported sadness (-1.47 vs -0.43; P = .0105), reduced sleep (-1.32 vs -0.21; P = .0388), reduced appetite (-0.93 vs -0.23; P = .0472), and lassitude (-0.82 vs 0.10; P = .0278). Decreases were significantly greater in the asenapine group compared with the olanzapine group for reported sadness (-1.47 vs -0.73; P = .0395), concentration difficulties (-1.69 vs -0.89; P = .0461), inability to feel (-1.18 vs -0.33; P = .0314), and pessimistic thoughts (-1.45 vs -0.53; P = .013). No statistically significant difference in decrease from baseline occurred in the olanzapine group compared with the placebo group.
Click figure to enlarge
By day 21, decreases from baseline were significantly greater in the asenapine group compared with the placebo group for reported sadness (-1.97 vs -0.93; P = .0318), inner tension (-1.50 vs -0.61; P = .0396), and inability to feel (-1.79 vs -0.80; P = .0324). Decreases were significantly greater in the asenapine group compared with the olanzapine group for inability to feel (-1.79 vs -0.90; P = .0354).
Effect on Manic Symptoms (YMRS)
The change (decrease) from baseline in LS mean YMRS total scores was greater in the asenapine group compared with the placebo and olanzapine groups at all time points assessed. Compared with placebo, these differences were significant on days 2 (-5.05 vs -1.61; P = .0131), 7 (-12.45 vs -6.12; P = .0249), 14 (-15.54 vs -7.66; P = .0032), and 21 (-16.58 vs -10.27; P = .0229).
Effect on SF-36 Component Scores
On day 21, change from baseline in the SF-36 physical component summary score was minimal in all groups (asenapine: −0.88; olanzapine: −1.53; placebo: −1.62) with no significant differences between groups. On day 21, there were also no significant differences in the SF-36 physical component subscale scores between the asenapine group and the olanzapine or placebo groups (Figure 2A).
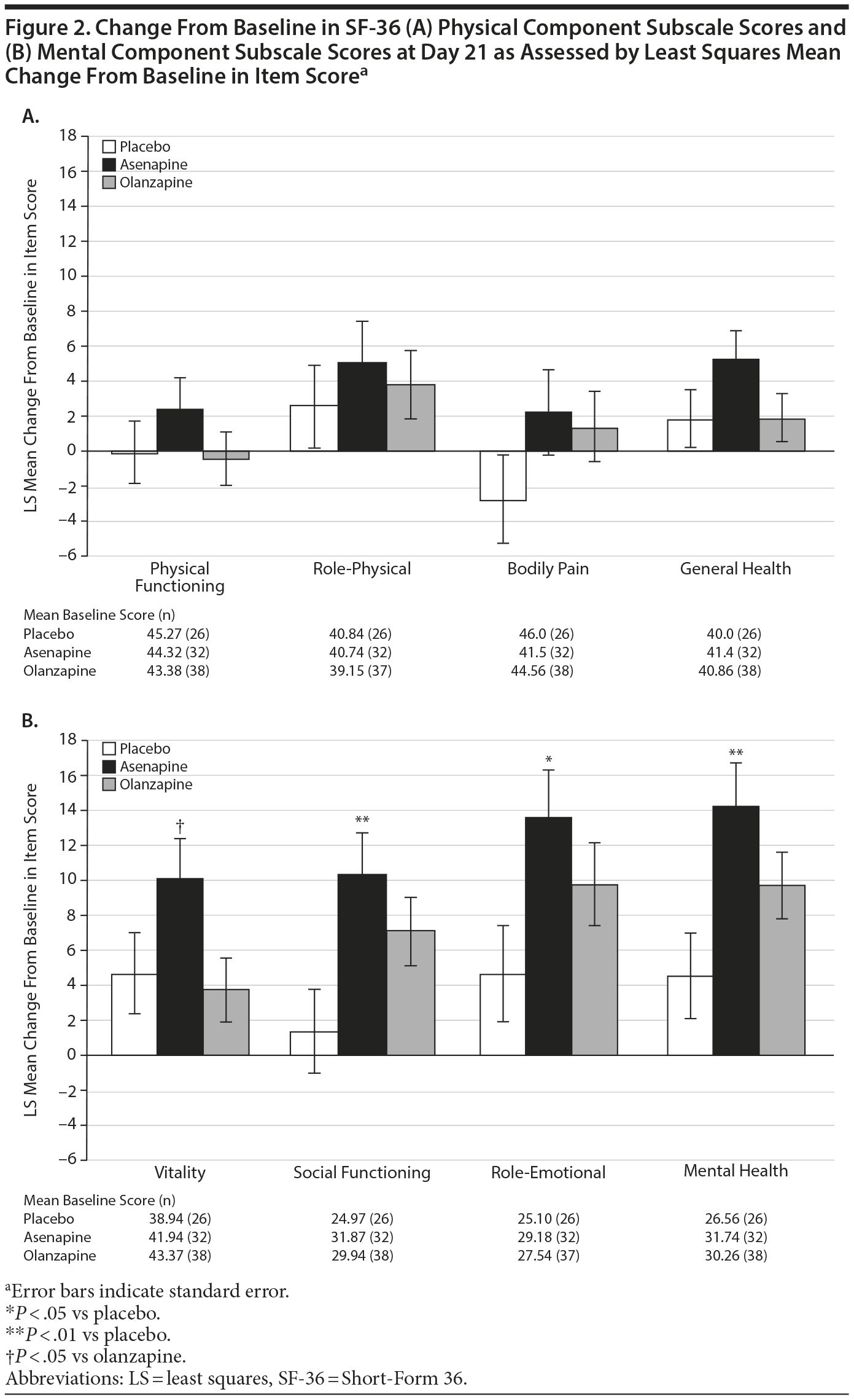
Click figure to enlarge
On day 21, change from baseline in the SF-36 mental component summary score was significantly greater in the asenapine group compared with the placebo group (16.57 vs 5.97; P = .0093). Change from baseline in this score in the olanzapine group compared with the placebo group was not significantly different (11.03 vs 5.97; P = .1501). In addition, on day 21, change from baseline in SF-36 mental component subscale scores was significantly greater in the asenapine group compared with the placebo and olanzapine groups (Figure 2B). The difference in improvement was statistically significant compared with placebo in the SF-36 subscales of social functioning (10.38 vs 1.32; P = .0093), role-emotional (13.58 vs 4.59; P = .0258), and mental health (14.28 vs 4.53; P = .0061). The difference in improvement was statistically significant compared with olanzapine in the SF-36 subscale of vitality (10.11 vs 3.70; P = .0311).
Adverse Events
In the asenapine group, adverse events that occurred in ≥ 5% of subjects and were more than twice as frequent as in the placebo group were increased appetite (asenapine 9.1%, placebo 0%), rash (9.1%, 3.9%), somnolence (9.1%, 3.9%), weight increase (9.1%, 0%), and akathisia, contusion, depression, hypoaesthesia oral, muscle twitching, pain in extremity, and tremor (all 6.1%, 0%; see Supplementary eTable 1 at PSYCHIATRIST.COM). In the olanzapine group, adverse events that occurred in ≥ 5% of subjects and were more than twice as frequent as in the placebo group were sedation (olanzapine 20.5%, placebo 7.7%), weight increase (15.4%, 0%), dry mouth (12.8%, 0%), and increased appetite, akathisia, tremor, and edema peripheral (all 5.1%, 0%).
DISCUSSION
This post hoc subgroup analysis demonstrated a significant effect on the primary outcome: decreases in MADRS scores in the asenapine group were significantly greater compared with the placebo group from baseline to day 7, day 21, and the endpoint. The reduction in MADRS scores in the asenapine group was significantly greater than that in the olanzapine group from baseline to day 7. Significant improvements in a number of individual depressive items were seen in the asenapine group, including greater improvement in reported sadness, sleep, appetite, and lassitude compared to placebo-treated individuals, and greater improvements were noted in the asenapine group compared with the olanzapine group for reported sadness, concentration difficulties, inability to feel, and pessimistic thoughts. Because of the hazards of uncorrected multiple comparisons regarding individual depressive symptoms, this finding should be seen as hypothesis generating rather than having any confirmative capacity. On secondary outcomes, significantly greater improvements from baseline to day 21 in the SF-36 mental component summary score were seen in the asenapine group but not the olanzapine compared with the placebo group. Significantly greater reductions in YMRS total scores in the asenapine group compared with the placebo group were evident on days 2, 7, and 21.
It is noteworthy that, as a class, atypical antipsychotics are, in general, effective in treating mania, but not all drugs in this class have shown efficacy in treating bipolar depression. Agents that show efficacy include quetiapine and olanzapine; however, other agents, such as ziprasidone and aripiprazole, have failed to meet primary efficacy endpoints.16,33 It is not yet clear if the results of these clinical trials reflect true differences in efficacy of various agents due to their unique pharmacologic profiles or to differences in study design. The latter has been suggested as a possibility given the high rate of placebo response observed in bipolar depression trials, which may have potentially obscured the antidepressant effects. If it is the former possibility of unique pharmacologic profiles, it is conceivable that the effects on α2-adrenergic and 5-HT7 receptors may confer asenapine treatment effects, although effects on other pathways may be involved.34
Limitations of the data include the modest sample size, differential dropout rates, and the pooling of the datasets of 2 randomized, double-blind source studies. These data, derived from a mixed cohort, cannot be directly compared with those from studies of patients with nonmixed major depression, and since the original studies were not designed for a major depressive episode analysis, these data need to be interpreted with caution as hypothesis generating rather than confirmatory. In addition, comparisons to olanzapine cannot be extrapolated to other atypical agents. In post hoc subgroup analyses, risk of type I errors (false positive) are potentially inflated because of multiple testing. Missing data can also lead to misinterpretation of results. To maximize the interpretability of these data, we aimed a priori to use an analytic plan that matched previous analyses as closely as possible.32
In summary, the findings from this analysis support the efficacy of asenapine in improving depressive symptoms in patients with bipolar I disorder who have moderate or severe major depressive episodes concurrently with a manic episode. The data suggest reduction in both mania and depression, a finding that raises the question of whether asenapine like quetiapine has bidirectional efficacy. In addition, asenapine was generally well tolerated in this subpopulation. The findings of this study warrant further investigation of the use of asenapine in treating bipolar depressive symptoms especially given the paucity of proven treatments for bipolar depressive symptoms.
Drug names: aripiprazole (Abilify), asenapine (Saphris), fluoxetine (Prozac and others), lithium (Lithobid and others), lurasidone (Latuda), olanzapine (Zyprexa), quetiapine (Seroquel), ziprasidone (Geodon).
Author affiliations: IMPACT Strategic Research Centre, Deakin University, Melbourne (Dr Berk); Department of Psychiatry, Orygen, The National Centre of Excellence in Youth Mental Health, and the Florey Institute for Neuroscience and Mental Health, University of Melbourne, Southbank VIC (Dr Berk); Department of Psychiatry, University of Melbourne, Albert Road Clinic, Melbourne (Dr Tiller), Australia; Merck, Rahway, New Jersey (Dr Zhao); UBC Department of Psychiatry, University of British Columbia, UBC Hospital, Vancouver, British Columbia, Canada (Dr Yatham); CADE Clinic, Discipline of Psychiatry, Sydney Medical School, University of Sydney, Sydney, Australia (Dr Malhi); and H. Lundbeck A/S, Copenhagen, Denmark (Dr Weiller).
Potential conflicts of interest: Dr Berk has received grant/research support from the National Institutes of Health, Cooperative Research Centre, Simons Autism Foundation, Cancer Council of Victoria, Stanley Medical Research Foundation, MBF, National Health and Medical Research Council (NHMRC), Beyond Blue, Rotary Health, Geelong Medical Research Foundation, Bristol-Myers Squibb, Eli Lilly, GlaxoSmithKline, Organon, Novartis, Mayne Pharma, Meat and Livestock Board, Servier and Woolworths; has been a speaker for Astra Zeneca, Bristol-Myers Squibb, Eli Lilly, GlaxoSmithKline, Janssen-Cilag, Lundbeck, Merck, Pfizer, Sanofi Synthelabo, Servier, Solvay, and Wyeth; and has served as a consultant to AstraZeneca, Bristol-Myers Squibb, Eli Lilly, GlaxoSmithKline, Janssen-Cilag, Lundbeck, Merck, and Servier. Dr Tiller has been a speaker for Lundbeck, Merck, AstraZeneca, Eli Lilly, Servier, and Janssen-Cilag and has served as a consultant to Lundbeck, Merck, AstraZeneca, Eli Lilly, and Servier. Dr Zhao was an employee of Merck when the research was conducted. Dr Yatham is on speaker/advisory boards for or has received research grants from AstraZeneca, Bristol Myers Squibb, Canadian Institutes of Health Research, Canadian Network for Mood and Anxiety Treatments, Eli Lilly, GlaxoSmithKline, Janssen, Lundbeck, the Michael Smith Foundation for Health Research, Pfizer, Servier, Sunovion, and the Stanley Foundation. Dr Malhi has received grant or research support from NHMRC, NSW Health, AstraZeneca, Eli Lilly, Organon, Pfizer, Servier, and Wyeth; has been a speaker for AstraZeneca, Eli Lilly, Janssen-Cilag, Lundbeck, Pfizer, Ranbaxy, Servier, and Wyeth; and has been a consultant for AstraZeneca, Eli Lilly, Janssen Cilag, Lundbeck, and Servier. Dr Weiller is an employee of H. Lundbeck A/S.
Funding/support: H. Lundbeck A/S provided statistical support and funded the medical writing support by CHC Group.
Role of the sponsor: H. Lundbeck A/S had no role in the conduct or publication of the study other than providing the writing support noted here.
Acknowledgments: Medical writing support was provided by Daniel McCallus, PhD; Arjen van Willigenburg, MSc; and Marc van Bekkum, MSc, at CHC Group; this support was funded by H. Lundbeck A/S. Dr McCallus, Mr van Willigenburg, and Mr. van Bekkum report no potential conflicts of interest relevant to the subject of this article.
Supplementary material: Available at PSYCHIATRIST.COM.
REFERENCES
1. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
2. Judd LL, Akiskal HS, Schettler PJ, et al. The long-term natural history of the weekly symptomatic status of bipolar I disorder. Arch Gen Psychiatry. 2002;59(6):530-537. PubMed doi:10.1001/archpsyc.59.6.530
3. Post RM, Denicoff KD, Leverich GS, et al. Morbidity in 258 bipolar outpatients followed for 1 year with daily prospective ratings on the NIMH life chart method. J Clin Psychiatry. 2003;64(6):680-690, quiz 738-739. PubMed doi:10.4088/JCP.v64n0610
4. Yatham LN, Lecrubier Y, Fieve RR, et al. Quality of life in patients with bipolar I depression: data from 920 patients. Bipolar Disord. 2004;6(5):379-385. PubMed doi:10.1111/j.1399-5618.2004.00134.x
5. Leverich GS, Altshuler LL, Frye MA, et al. Factors associated with suicide attempts in 648 patients with bipolar disorder in the Stanley Foundation Bipolar Network. J Clin Psychiatry. 2003;64(5):506-515. PubMed doi:10.4088/JCP.v64n0503
6. Sato T, Bottlender R, Schröter A, et al. Frequency of manic symptoms during a depressive episode and unipolar ‘ depressive mixed state’ as bipolar spectrum. Acta Psychiatr Scand. 2003;107(4):268-274. PubMed doi:10.1034/j.1600-0447.2003.00051.x
7. Malhi GS, Bargh DM, Kuiper S, et al. Modeling bipolar disorder suicidality. Bipolar Disord. 2013;15(5):559-574. PubMed doi:10.1111/bdi.12093
8. Berk M, Berk L, Dodd S, et al. The sick role, illness cognitions and outcomes in bipolar disorder. J Affect Disord. 2013;146(1):146-149. PubMed doi:10.1016/j.jad.2012.07.003
9. Stange JP, Sylvia LG, da Silva Magalh×£es PV, et al. Extreme attributions predict the course of bipolar depression: results from the STEP-BD randomized controlled trial of psychosocial treatment. J Clin Psychiatry. 2013;74(3):249-255. PubMed doi:10.4088/JCP.12m08019
10. Barnett JH, Smoller JW. The genetics of bipolar disorder. Neuroscience. 2009;164(1):331-343. PubMed doi:10.1016/j.neuroscience.2009.03.080
11. Lagerberg TV, Sundet K, Aminoff SR, et al. Excessive cannabis use is associated with earlier age at onset in bipolar disorder. Eur Arch Psychiatry Clin Neurosci. 2011;261(6):397-405. PubMed doi:10.1007/s00406-011-0188-4
12. Larsson S, Aas M, Klungs׸yr O, et al. Patterns of childhood adverse events are associated with clinical characteristics of bipolar disorder. BMC Psychiatry. 2013;13(1):97. PubMed doi:10.1186/1471-244X-13-97
13. Price AL, Marzani-Nissen GR. Bipolar disorders: a review. Am Fam Physician. 2012;85(5):483-493. PubMed
14. Mart×nez-Arán A, Vieta E, Reinares M, et al. Cognitive function across manic or hypomanic, depressed, and euthymic states in bipolar disorder. Am J Psychiatry. 2004;161(2):262-270. PubMed doi:10.1176/appi.ajp.161.2.262
15. Colom F, Vieta E, Martinez-Aran A, et al. A randomized trial on the efficacy of group psychoeducation in the prophylaxis of recurrences in bipolar patients whose disease is in remission. Arch Gen Psychiatry. 2003;60(4):402-407. PubMed doi:10.1001/archpsyc.60.4.402
16. Yatham LN, Kennedy SH, Parikh SV, et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) and International Society for Bipolar Disorders (ISBD) collaborative update of CANMAT guidelines for the management of patients with bipolar disorder: update 2013. Bipolar Disord. 2013;15(1):1-44. PubMed doi:10.1111/bdi.12025
17. Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry. 2001;158(6):906-912. PubMed doi:10.1176/appi.ajp.158.6.906
18. Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med. 2007;356(17):1711-1722. PubMed doi:10.1056/NEJMoa064135
19. Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry. 2003;60(11):1079-1088. PubMed doi:10.1001/archpsyc.60.11.1079
20. Lombardo I, Sachs G, Kolluri S, et al. Two 6-week, randomized, double-blind, placebo-controlled studies of ziprasidone in outpatients with bipolar I depression: did baseline characteristics impact trial outcome? J Clin Psychopharmacol. 2012;32(4):470-478. PubMed doi:10.1097/JCP.0b013e31825ccde5
21. Thase ME, Jonas A, Khan A, et al. Aripiprazole monotherapy in nonpsychotic bipolar I depression: results of 2 randomized, placebo-controlled studies. J Clin Psychopharmacol. 2008;28(1):13-20. PubMed doi:10.1097/jcp.0b013e3181618eb4
22. Loebel A, Cucchiaro J, Silva R, et al. Lurasidone monotherapy in the treatment of bipolar I depression: a randomized, double-blind, placebo-controlled study. Am J Psychiatry. 2014;171(2):160-168. PubMed doi:10.1176/appi.ajp.2013.13070984
23. Loebel A, Cucchiaro J, Silva R, et al. Lurasidone as adjunctive therapy with lithium or valproate for the treatment of bipolar I depression: a randomized, double-blind, placebo-controlled study. Am J Psychiatry. 2014;171(2):169-177. PubMed doi:10.1176/appi.ajp.2013.13070985
24. Judd LL, Akiskal HS. The prevalence and disability of bipolar spectrum disorders in the US population: re-analysis of the ECA database taking into account subthreshold cases. J Affect Disord. 2003;73(1-2):123-131. PubMed doi:10.1016/S0165-0327(02)00332-4
25. Berk M, Berk L, Davey CG, et al. Treatment of bipolar depression. MJA Open. 2012;1(suppl 4):32-35.
26. McIntyre RS, Cohen M, Zhao J, et al. A 3-week, randomized, placebo-controlled trial of asenapine in the treatment of acute mania in bipolar mania and mixed states. Bipolar Disord. 2009;11(7):673-686. PubMed doi:10.1111/j.1399-5618.2009.00748.x
27. McIntyre RS, Cohen M, Zhao J, et al. Asenapine in the treatment of acute mania in bipolar I disorder: a randomized, double-blind, placebo-controlled trial. J Affect Disord. 2010;122(1-2):27-38. PubMed doi:10.1016/j.jad.2009.12.028
28. Young RC, Biggs JT, Ziegler VE, et al. A rating scale for mania: reliability, validity and sensitivity. Br J Psychiatry. 1978;133(5):429-435. PubMed doi:10.1192/bjp.133.5.429
29. Sheehan DV, Lecrubier Y, Sheehan KH, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59(suppl 20):22-33, quiz 34-57. PubMed
30. Montgomery SM. Depressive symptoms in acute schizophrenia. Prog Neuropsychopharmacol. 1979;3(4):429-433. PubMed doi:10.1016/0364-7722(79)90058-4
31. Müller-Thomsen T, Arlt S, Mann U, et al. Detecting depression in Alzheimer’s disease: evaluation of four different scales. Arch Clin Neuropsychol. 2005;20(2):271-276. PubMed doi:10.1016/j.acn.2004.03.010
32. Thase ME, Macfadden W, Weisler RH, et al; BOLDER II Study Group. Efficacy of quetiapine monotherapy in bipolar I and II depression: a double-blind, placebo-controlled study (the BOLDER II study). J Clin Psychopharmacol. 2006;26(6):600-609. PubMed doi:10.1097/01.jcp.0000248603.76231.b7
33. Malhi GS, Bargh DM, McIntyre R, et al. Balanced efficacy, safety, and tolerability recommendations for the clinical management of bipolar disorder. Bipolar Disord. 2012;14(suppl 2):1-21. PubMed doi:10.1111/j.1399-5618.2012.00989.x
34. Grossini E, Gramaglia C, Farruggio S, et al. Asenapine increases nitric oxide release and protects porcine coronary artery endothelial cells against peroxidation. Vascul Pharmacol. 2014;60(3):127-141. PubMed doi:10.1016/j.vph.2014.01.008
Download Free PDF
This PDF is free for all visitors!
![]() Save
Save
![]() Share
Share
![]() Cite
Cite