Objective: This phase 3, randomized, double-blind, placebo-controlled study evaluated the efficacy and tolerability of fixed-dose levomilnacipran sustained release (SR) compared with placebo in patients with major depressive disorder (MDD); the study was conducted from September 2009-May 2011.
Method: Outpatients met DSM-IV-TR criteria for MDD with an ongoing major depressive episode ≥ 8 weeks’ duration. After a 1-week placebo lead-in, patients were randomly assigned to receive placebo (n = 179) or levomilnacipran SR 40 mg (n = 181), 80 mg (n = 181), or 120 mg (n = 183) once daily for 8 weeks of double-blind treatment, followed by a 2-week double-blind down-taper. The primary efficacy parameter was change from baseline on the clinician-rated Montgomery-Asberg Depression Rating Scale (MADRS) total score. The prespecified secondary efficacy parameter was change from baseline in Sheehan Disability Scale (SDS) total score. Additional efficacy measures included the 17-item Hamilton Depression Rating Scale (HDRS17) and Clinical Global Impressions-Severity of Illness (CGI-S) and -Improvement (CGI-I). Safety and tolerability were also evaluated.
Results: The least squares mean difference (LSMD) for change from baseline in MADRS total score was significantly superior to placebo for all dose groups: −3.23 (P = .0186), −3.99 (P = .0038), and −4.86 (P = .0005) for levomilnacipran SR 40, 80, and 120 mg, respectively. The LSMD was significantly different for levomilnacipran SR 80 mg and 120 mg versus placebo on the SDS (−2.51 and −2.57, respectively, P < .05 for both doses), HDRS17 (−2.09 and −2.34, respectively, P < .05 for both doses), CGI-S (−0.43 [P < .01] and −0.35 [P < .05], respectively), and CGI-I (−0.34 and −0.32, respectively, P < .05 for both doses) assessments. The most common treatment-emergent adverse events (≥ 10% of any treatment group) were headache, nausea, constipation, dry mouth, increased heart rate, and hyperhidrosis.
Conclusions: Levomilnacipran SR demonstrated significant improvement in depressive symptoms and functioning relative to placebo. In this study, levomilnacipran SR was generally well tolerated.
Trial Registration: ClinicalTrials.gov identifier: NCT00969709
J Clin Psychiatry 2013;74(3):242-248
© Copyright 2013 Physicians Postgraduate Press, Inc.
Submitted: September 28, 2012; accepted January 28, 2013 (doi:10.4088/JCP.12m08197).
Corresponding author: Gregory M. Asnis, MD, Albert Einstein College of Medicine, Montefiore Medical Center, 111 East 210th St, Bronx, NY 10467 ([email protected]).
Efficacy and Safety of Levomilnacipran Sustained Release
40 mg, 80 mg, or 120 mg in Major Depressive Disorder:
A Phase 3, Randomized, Double-Blind, Placebo-Controlled Study
ABSTRACT
Objective: This phase 3, randomized, double-blind,
placebo-controlled study evaluated the efficacy and tolerability of fixed-dose levomilnacipran sustained
release (SR) compared with placebo in patients with major depressive disorder (MDD); the study was conducted from September 2009–May 2011.
Method: Outpatients met DSM-IV-TR criteria for MDD with an ongoing major depressive episode ≥ 8 weeks’ duration. After a 1-week placebo lead-in, patients were randomly assigned to receive placebo (n = 179) or levomilnacipran SR 40 mg (n = 181), 80 mg (n = 181), or 120 mg (n = 183) once daily for 8 weeks of double-blind treatment, followed by a 2-week double-blind down-taper. The primary efficacy parameter was change from baseline on the clinician-rated Montgomery-Asberg Depression Rating Scale (MADRS) total score. The prespecified secondary efficacy parameter was change from baseline in Sheehan Disability Scale (SDS) total score. Additional efficacy measures included the 17-item Hamilton Depression Rating Scale (HDRS17) and Clinical Global Impressions-Severity of Illness (CGI-S) and -Improvement (CGI-I). Safety and tolerability were
also evaluated.
Results: The least squares mean difference (LSMD) for change from baseline in MADRS total score was significantly superior to placebo for all dose groups: −3.23 (P = .0186), −3.99 (P = .0038), and −4.86 (P = .0005) for levomilnacipran SR 40, 80, and 120 mg, respectively. The LSMD was significantly different for levomilnacipran SR 80 mg and 120 mg versus placebo on the SDS (−2.51 and −2.57, respectively, P < .05 for both doses), HDRS17 (−2.09 and −2.34, respectively, P < .05 for both doses), CGI-S (−0.43 [P < .01] and −0.35 [P < .05], respectively), and CGI-I (−0.34 and −0.32, respectively, P < .05 for both doses) assessments. The most common treatment-emergent adverse events (≥ 10% of any treatment group) were headache, nausea, constipation, dry mouth, increased heart rate, and hyperhidrosis.
Conclusions: Levomilnacipran SR demonstrated significant improvement in depressive symptoms and functioning relative to placebo. In this study, levomilnacipran SR was generally well tolerated.
Trial Registration: ClinicalTrials.gov identifier: NCT00969709
J Clin Psychiatry 2013;74(3):242–248
© Copyright 2013 Physicians Postgraduate Press, Inc.
Submitted: September 28, 2012; accepted January 28, 2013 (doi:10.4088/JCP.12m08197).
Corresponding author: Gregory M. Asnis, MD, Albert Einstein College of Medicine, Montefiore Medical Center, 111 East 210th St, Bronx, NY 10467 ([email protected]).
The complex nature of major depressive disorder (MDD) suggests that recovery may be most appropriately judged by multiple factors. Even when patients achieve symptom improvement, impaired social and occupational functioning may persist and interfere with well-being. As such, it has been suggested that return to wellness in patients with MDD may be better defined by evaluating a combination of symptoms, functional status, and pathophysiologic changes.1 The development of effective and safe new medications that address all aspects of MDD treatment is essential.
Levomilnacipran (1S, 2R-milnacipran) is a potent and selective serotonin-norepinephrine reuptake inhibitor (SNRI) in late-stage clinical development for treatment of MDD in adults. A sustained release (SR) formulation of levomilnacipran was developed to
allow for once-daily dosing. In vitro studies have shown that levomilnacipran has approximately 2-fold greater potency for norepinephrine relative to serotonin reuptake inhibition; levomilnacipran shows over 10-fold higher selectivity for norepinephrine versus serotonin reuptake inhibition compared with duloxetine or venlafaxine.2
In addition to this phase 3 study (ClinicalTrials.gov identifier:
NCT00969709), the safety and efficacy of levomilnacipran SR in
the treatment of MDD have been evaluated in 4 fixed- and
flexible-dose randomized, double-blind, placebo-controlled trials (ClinicalTrials.gov identifiers NCT01377194, NCT01034462, and NCT00969150 and EudraCT number 2006-002404-34). The current study was designed as a fixed-dose study to evaluate the
safety and efficacy of levomilnacipran SR (40, 80, or 120 mg/d) relative to placebo in the treatment of adult patients with MDD.
METHOD
This study was conducted at 38 US study centers between
September 2009 and May 2011 in full compliance with US Food and Drug Administration guidelines for Good Clinical Practice and the ethical principles of the Declaration of Helsinki. The study protocol was approved by each site’s institutional review board, and all patients provided written informed consent.
Study Design
This study was an 11-week multicenter, randomized, double-blind, placebo-controlled, parallel-group, fixed-dose study conducted in outpatients with MDD to evaluate the efficacy,
safety, and tolerability of fixed-dose levomilnacipran SR compared with placebo. The study comprised a 1-week, single-blind, placebo run-in period, followed by 8-week double-blind treatment and a 2-week double-blind down-taper period. Eligible patients were randomly assigned on a 1:1:1:1 basis to placebo or once-daily levomilnacipran SR 40 mg, 80 mg, or 120 mg. Levomilnacipran SR was initiated at 20 mg/d, and doses were increased to 40 mg/d on day 2; the 80-mg/d and 120-mg/d target doses were reached on day 5 and day 8, respectively.
Patients were randomized by a computer-generated list of numbers and assigned to identically appearing treatment. Investigators and patients were blinded to allocation of the investigational product throughout treatment and down-taper periods. The blind was maintained via a secured randomization code list and was broken only in case of emergency; unblinding disqualified a patient from further study participation.
Inclusion Criteria
Male or female patients (18–65 years of age, inclusive) who met criteria for MDD as defined by the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR),3 with the diagnosis confirmed by the Mini-International Neuropsychiatric Interview,4 participated in the study. Patients were required to have a current ongoing depressive episode ≥ 8 weeks’ duration, score ≥ 30 on the clinician-rated Montgomery-Asberg Depression Rating Scale (MADRS)5 at screening and baseline, score ≥ 26 on the self-rated MADRS (MADRS-SR) at baseline, body mass index ≥ 18 and ≤ 40, and negative pregnancy test results.
Exclusion Criteria
Patients with clinically significant abnormalities on physical examination, clinical laboratory tests, or electrocardiography (ECG) were excluded. Patients with DSM-IV-TR primary Axis I diagnoses other than MDD, lifetime history of manic/hypomanic episode, other significant psychiatric disorders, or substance abuse/dependence within 6 months of the study were excluded. Patients with significant medical conditions (eg, central nervous system disorders, cardiovascular diseases, clinically significant systolic and/or diastolic blood pressure readings) or suicide risk (ie, suicide attempt within the past year, score ≥ 5 on MADRS item 10 [suicidal thoughts] or significant risk based on investigator judgment or Columbia–Suicide Severity Rating Scale [C-SSRS]6 information) were ineligible. Patients with a history of intolerance or hypersensitivity to milnacipran, other SNRIs, or selective serotonin reuptake inhibitors (SSRIs) or nonresponse to ≥ 2 antidepressants after treatment with adequate dose and duration were excluded. Patients taking concomitant psychoactive medications (with the exception of eszopiclone, zolpidem, or zaleplon for insomnia) were also excluded.
Efficacy and Safety Assessments
The primary and secondary efficacy assessments were the MADRS (screening [week −1], baseline [week 0], weeks 1, 2, 4, 6, 8) and the Sheehan Disability Scale (SDS)7 (weeks 0, 4, 6, 8).
Additional efficacy measures included the 17-item
Hamilton Depression Rating Scale (HDRS17)8 (weeks −1, 0, 1, 2, 4, 6, 8) and the Clinical Global Impressions-Severity of Illness (CGI-S) (weeks 0, 1, 2, 4, 6, 8) and -Improvement (CGI-I) (weeks 1, 2, 4, 6, 8).9
Adverse events (AEs) were assessed at all double-blind study visits (weeks 0, 1, 2, 4, 6, and 8 and down-taper period) and evaluated by intensity (mild, moderate, or severe) and possible relationship to study drug. At each study visit, patients were queried about AEs that may have occurred since the previous visit, and AEs were recorded using preferred terms based on MedDRA coding of investigator terms for each event; no specific AE scales were utilized. Clinical laboratory tests (weeks −1, 4, 8 or early termination), vital signs (weeks −1, 0, 1, 2, 4, 6, 8), and 12-lead ECGs (weeks −1, 4, 8) were evaluated. The C-SSRS (weeks −1, 0, 1, 2, 4, 6, 8) assessed the severity of suicidal behavior and ideation.
Statistical Analyses
The safety population comprised randomized patients who received ≥ 1 dose of double-blind study medication; the modified intent-to-treat (ITT) population was defined as all patients in the safety population with ≥ 1 postbaseline MADRS total score.
Analysis of the prespecified primary efficacy parameter, MADRS total score change from baseline to week 8, was performed on the modified ITT population using a mixed-effects model for repeated measures (MMRM) approach with treatment group, pooled study center, visit, and treatment group–by-visit interaction as fixed effects and the baseline MADRS and baseline-by-visit interaction as covariate. Primary comparisons were between each levomilnacipran SR dose group versus placebo at week 8. To control for potential type I error rate resulting from testing multiple comparisons, the Hochberg procedure10 was used. Sensitivity analyses using an analysis of covariance (ANCOVA) model and
pattern-mixture model (PMM) approaches were performed
to assess the robustness of the primary results. The ANCOVA model included treatment group and pooled study center as factors and baseline MADRS total score as covariate, with missing data imputed using the last-observation-carried-forward (LOCF) method. The PMM approach was based on nonfuture-dependent missing value restrictions.11
The secondary efficacy parameter was change from baseline to week 8 in SDS total score. SDS total score was calculated using only patients with valid responses on all 3 subscale scores; if 1 or more subscales were missing, the SDS total score was set equal to missing. Statistical analysis was similar to the primary efficacy parameter.
Additional efficacy parameters included change from baseline to week 8 in SDS subscale scores, HDRS17 total score, CGI-S score, MADRS response rate (≥ 50% improvement from baseline), MADRS remission rate (total score ≤ 10), and CGI-I score at week 8. All statistical tests were 2-sided hypothesis tests performed at the 5% level of significance; confidence intervals (CIs) were 2-sided 95% CIs.
Safety analyses were performed for the
double-blind and down-taper periods using the safety population; for each parameter, the last assessment before the first dose of double-blind study medication was used as baseline. Statistical analysis for demographic characteristics was
analyzed by ANOVA (continuous variables) or Cochran-Mantel-Haenszel test (categorical variables).12 Between-group comparisons for overall and by-reason discontinuations were performed using a Fisher exact test.13
RESULTS
Patient Disposition and
Demographic Characteristics
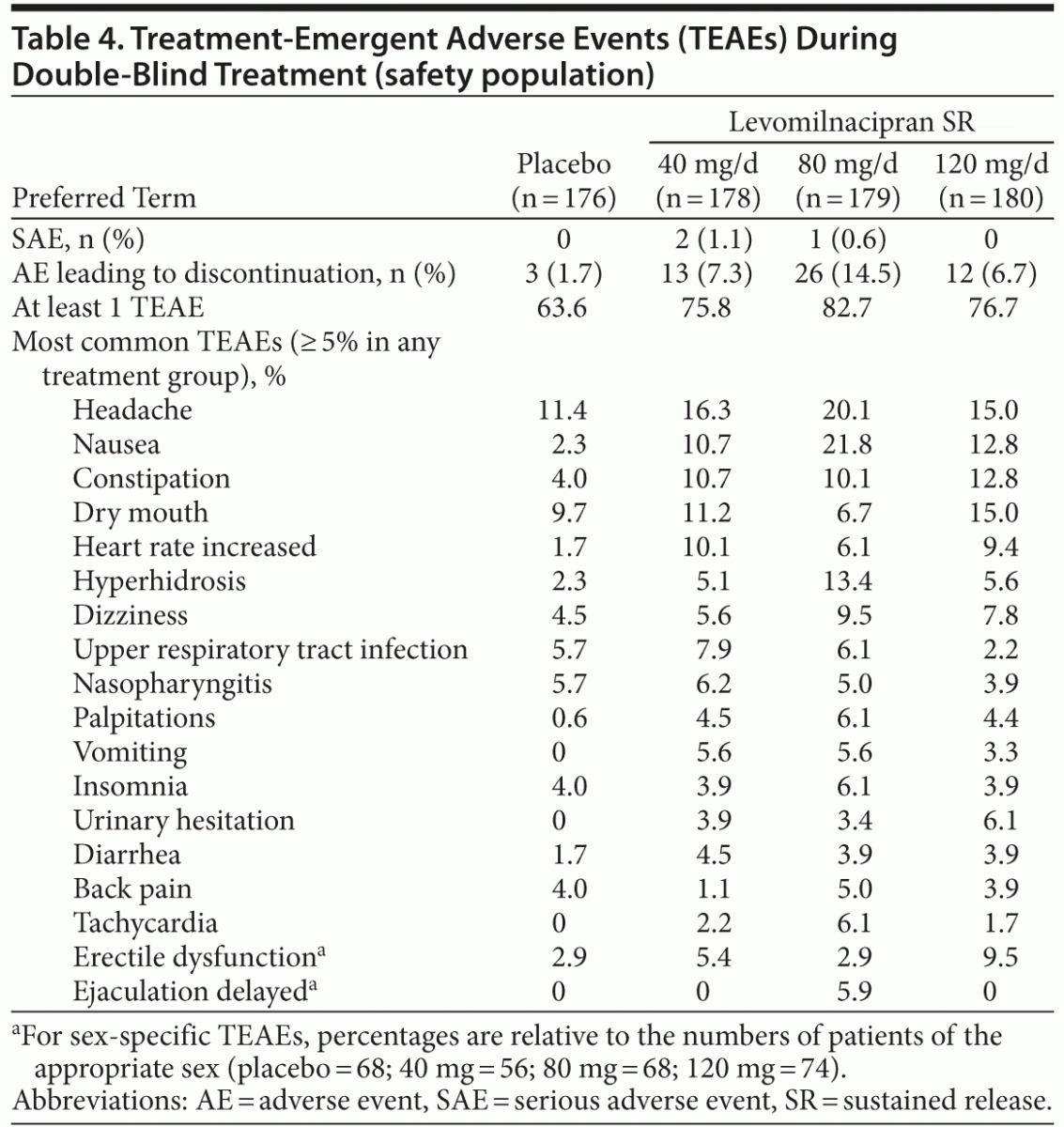
A total of 724 patients were randomized to receive double-blind treatment; there were 713 patients in the safety population and 704 patients in the modified ITT population. Reasons for premature discontinuation are presented in Table 1. Significantly more levomilnacipran SR than placebo patients discontinued due to AEs (40 mg: P = .0185, 80 mg: P ≤ .001, 120 mg: P = .0316). The common AEs that led to discontinuation were nausea (placebo: 0, 40 mg: 1.1%, 80 mg: 3.4%, 120 mg: 0), vomiting (placebo: 0, 40 mg: 0.6%, 80 mg: 1.7%, 120 mg: 0), and palpitations (placebo: 0, 40 mg: 0, 80 mg: 1.7%, 120 mg: 0).
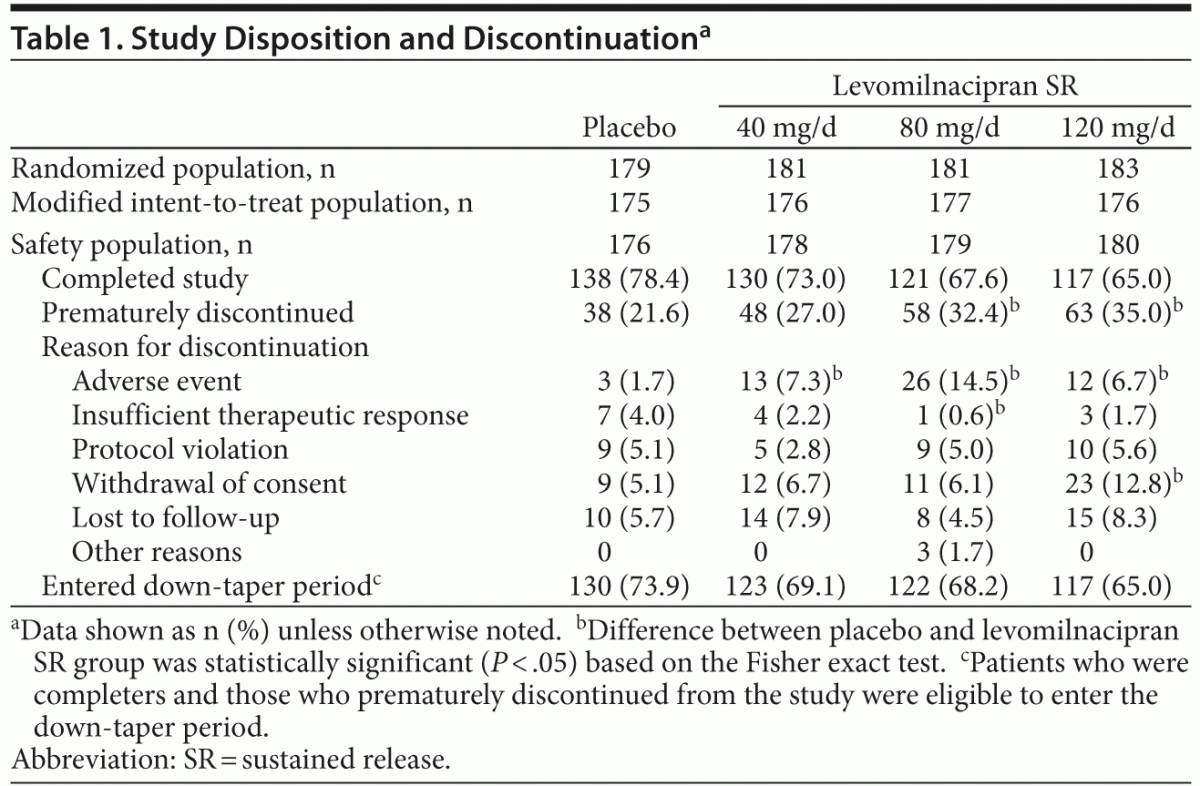
There were no relevant differences between treatment groups for baseline demographic characteristics or depression history (Table 2). The mean baseline MADRS score (36) exceeded the cutoff score used as the threshold to define severe depression.14 Most patients (76%) had a history of recurrent depression, and the mean duration of illness was approximately 11 years. Approximately half of all patients had received prior antidepressant therapy within 5 years of the screening visit.
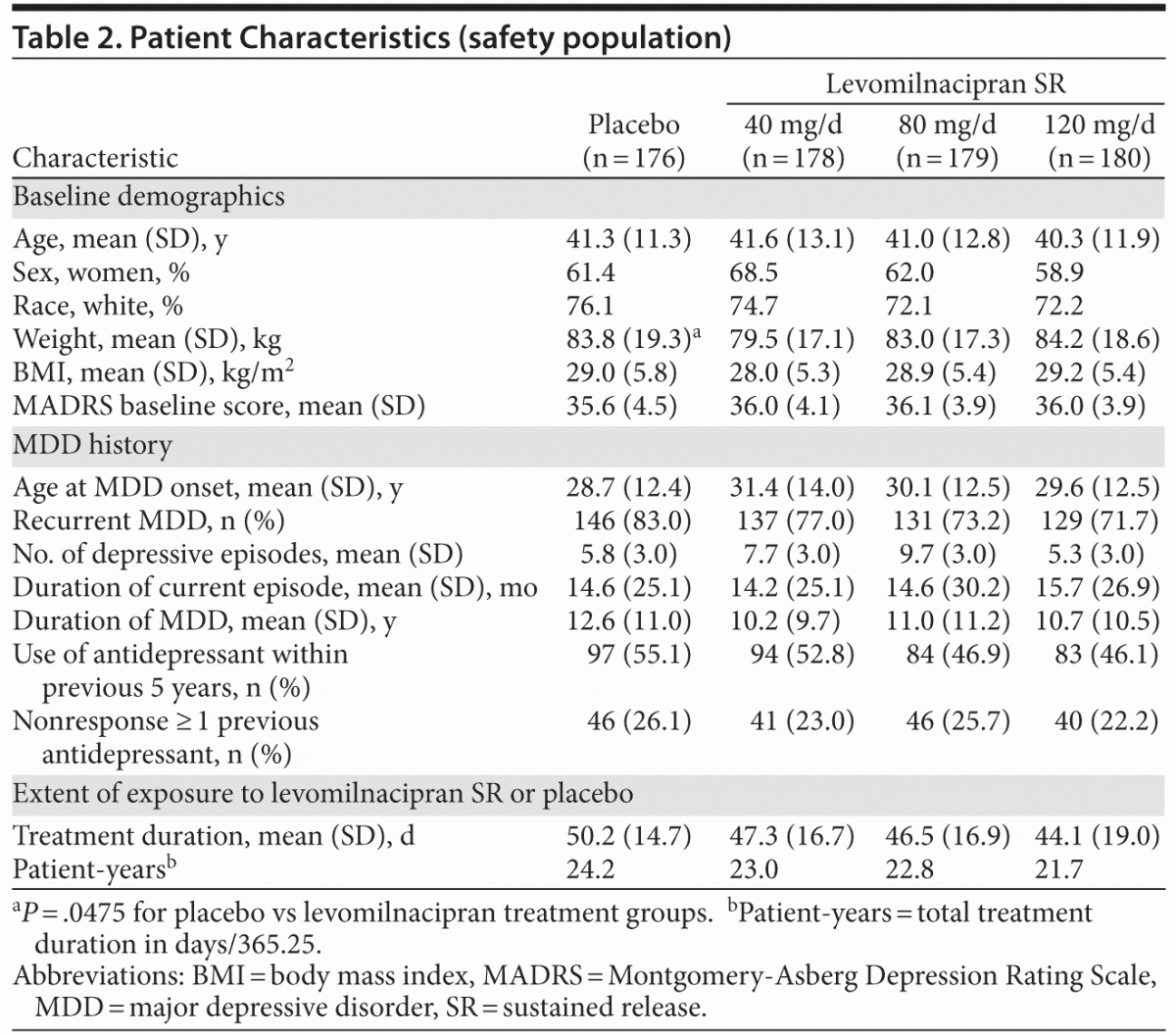
Efficacy
Significant improvement was seen in each dose group relative to placebo following a closed testing procedure accounting for multiplicity. Least squares (LS) mean change in MADRS total score at the end of week 8 (MMRM) was −14.8 for the levomilnacipran SR 40-mg group, −15.6 for the 80-mg group, and −16.5 for the 120-mg group compared with −11.6 for placebo; significant advantage over placebo was observed by week 4 in the 80-mg and 120-mg groups (Figure 1). LOCF and PMM sensitivity analyses on MADRS change from baseline supported the primary analysis. The LS mean change was −10.7 for placebo, −13.3 for the 40-mg group (P = .0410), −14.1 for the 80-mg group (P = .0058), and −14.1 for the 120-mg group (P = .0063) at the end of week 8 (LOCF). For all selected values of the shift parameter in the PMM analysis, mean changes in MADRS total
score remained significantly greater in levomilnacipran SR–treated patients than in placebo-treated patients. Results remained significant for both MMRM and LOCF analyses after adjustment for multiple comparisons. Change from baseline to week 8 on the secondary efficacy measure, SDS total score, was significantly greater for levomilnacipran SR 80 and 120 mg/d versus placebo (Table 3).
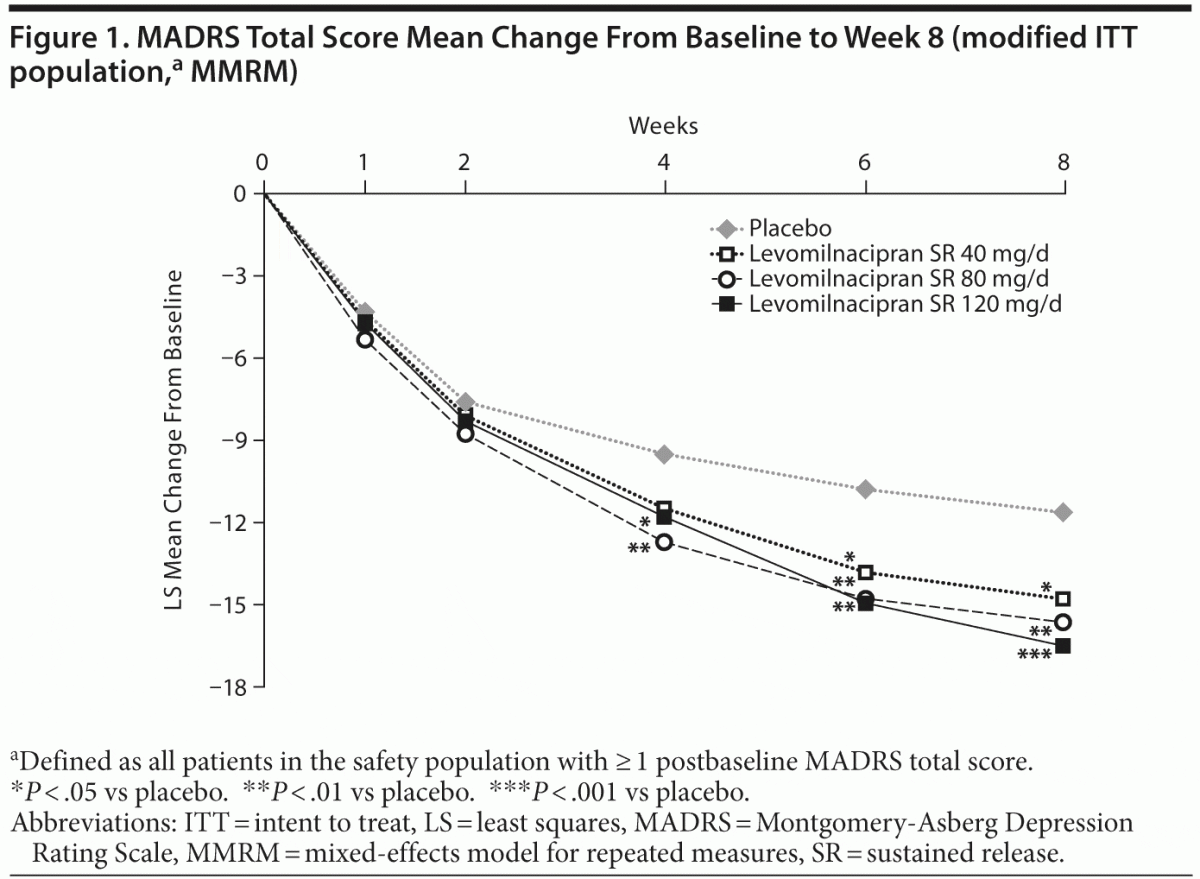
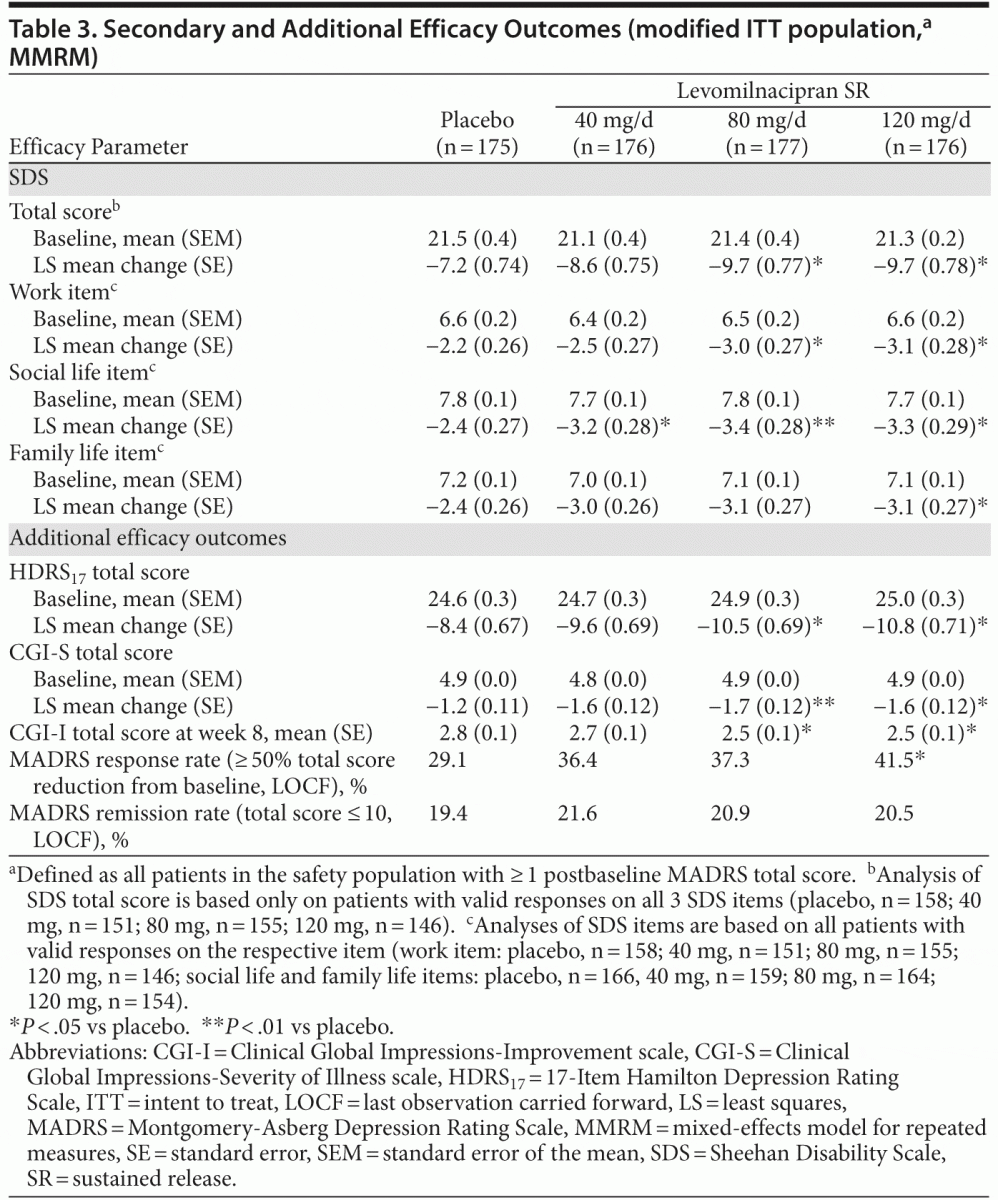
Significant improvements versus placebo were also consistently observed on additional efficacy parameters for
higher doses of levomilnacipran SR (Table 3). MADRS response rate (≥ 50% total score reduction) was significantly higher for levomilnacipran SR 120-mg patients (41.5%) compared with placebo (29.1%; P = .0107); there was no statistically significant difference in MADRS remission rates (total score ≤ 10) for any levomilnacipran group relative to placebo.
Safety and Tolerability
The mean duration of double-blind treatment ranged from 44 to 50 days across groups. No deaths were reported in this study. An overall summary of AEs and the most common (≥ 5% in any treatment group) TEAEs is presented in Table 4. Most TEAEs were considered by the investigator to be mild or moderate in intensity. During double-blind treatment, serious AEs (SAEs) were reported in 2 patients (1.1%) in the levomilnacipran SR 40-mg group (chest pain and deep vein thrombosis in 1 patient and aggression in 1 patient) and 1 patient (0.6%) in the 80-mg group (cytomegalovirus mononucleosis). During double-blind down-taper, approximately 9% of placebo-treated patients and 7%–9% of levomilnacipran SR–treated patients had a newly emergent AE. Nasopharyngitis was the most frequently reported AE during down-taper with an incidence greater than placebo (0 patients in the placebo group; 3 patients in the 40-mg group; 1 each in the 80-mg and 120-mg groups).
During double-blind treatment, slight mean (SD) increases in aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were observed in all levomilnacipran SR dose groups relative to placebo (AST = 0.5 [6.5] U/L; ALT = 1.4 [9.3] U/L); increases were more pronounced in the 80-mg dose group (AST = 4.4 [28.0] U/L; ALT = 5.4 [32.5] U/L) than in the 40-mg (AST = 1.6 [16.0] U/L; ALT = 2.2 [15.2] U/L) or 120-mg (AST = 3.4 [30.3] U/L; ALT = 1.6 [14.5] U/L) groups. These increases were largely due to 7 levomilnacipran SR patients (2 patients in the 40-mg group, 2 patients in the 80-mg group, and 3 patients in the 120-mg group) who had postbaseline ALT and/or AST values that met potentially clinically significant (PCS) criteria (≥ 3 × upper limit of normal [ULN]). Creatine kinase levels were not obtained in this study, so the organ sources of these transaminase elevations were not established. No patients met the criteria for Hy’s law15 (ALT or AST elevation ≥ 3 × ULN, total bilirubin elevation > 2 × ULN, and alkaline phosphatase < 2 × ULN). Mean changes in other chemistry, hematologic, and urinalysis laboratory measures and incidence of PCS changes were small and similar among all treatment groups.
Levomilnacipran SR was weight neutral during the study; mean (SD) change in body weight at the end of double-blind treatment was small and similar across groups (placebo, +0.19 [2.06] kg; levomilnacipran SR 40 mg, –0.50 [1.83] kg; 80 mg, –0.77 [2.04] kg; 120 mg, –0.75 [2.06] kg. Mean (SD) increases in supine pulse rate were greater for levomilnacipran SR (9.1 [10.5] bpm, 8.6 [12.6] bpm, and 9.1 [10.6] bpm for levomilnacipran SR 40 mg, 80 mg, and 120 mg, respectively) than for placebo (0.5 [8.7] bpm). Mean (SD) change in systolic blood pressure from baseline to end of double-blind treatment was 0.9 (10.6) mm Hg for placebo and 2.7 (10.4) mm Hg, 4.7 (10.5) mm Hg, and 2.7 (8.9) mm Hg for levomilnacipran SR 40 mg, 80 mg, and 120 mg, respectively. Mean (SD) change in diastolic blood pressure was −0.1 (7.9) mm Hg for placebo and 2.7 (8.1) mm Hg, 3.8 (7.8) mm Hg, and 2.6 (7.1) mm Hg for levomilnacipran SR 40 mg, 80 mg, and 120 mg, respectively.
Mean increases in QTcF interval were not seen in any treatment group, and no patients met
QTcF PCS criteria (interval > 500 msec). Mean (SD) increases in QTcB interval were greater in the levomilnacipran SR groups (7.5 [22.9] msec, 7.3 [19.9] msec, and 10.5 [21.3] msec for the 40-, 80-, and 120-mg groups, respectively) compared with placebo (0.5 [20.2] msec); these increases were consistent with the increases in ventricular heart rate.
The incidence of suicidal ideation as assessed by the C-SSRS was similar in the placebo and levomilnacipran SR 80-mg groups (approximately 31% in both groups) and slightly lower in the 40-mg and 120-mg groups (27% in both groups). One placebo patient (0.6%), 2 levomilnacipran SR 40-mg patients (1.1%), and 2 levomilnacipran SR 120-mg patients (1.1%) reported C-SSRS suicidal behavior.
Incidences of TEAEs related to suicidal ideation or behavior were low and similar between groups during double-blind treatment (suicidal ideation: 1 placebo, levomilnacipran SR 40-mg, and levomilnacipran SR 80-mg patient each; suicidal behavior: 1 placebo and levomilnacipran SR 120-mg patient each). One patient in the levomilnacipran SR 40-mg group had an SAE of suicide attempt during the down-taper period (10 days after stop of double-blind treatment); it was considered by the investigator to be severe and not related to study drug. Although this patient reported a history of 2 prior suicide attempts and 1 other aborted suicide attempt, she was not evaluated as a current suicide risk at the time of study entry.
DISCUSSION
In this phase 3 fixed-dose study, robust efficacy was demonstrated by significant change in MADRS total score (MMRM) in favor of levomilnacipran SR versus placebo (LSMD: 40 mg/d = −3.23, 80 mg/d = −3.99, and 120 mg/d = −4.86). Higher doses produced numerically greater change, and significant separation from placebo occurred earlier in the 80-mg and 120-mg dose groups than in the 40-mg group.
Significant differences versus placebo were consistently observed across secondary and additional efficacy measures in higher-dose groups. Improvement in SDS total score was noted in all levomilnacipran groups versus placebo at week 8; the difference was statistically significant versus placebo at the 80-mg and 120-mg doses. Levomilnacipran SR 120 mg produced significant improvement versus placebo on all of the SDS subscales. Research suggests that the MADRS and SDS are both sensitive to treatment effects16 and that they measure independent symptom and functional domains.17
In short-term studies, an average 2-point difference on the MADRS is frequently used as the standard to establish that treatment effects are clinically relevant.18 In the present study, MADRS effect size exceeds the 2-point standard for all levomilnacipran SR doses, with a treatment effect ≥ 4 points in the higher dose groups.
Response rate is also frequently used as a measure of clinical relevance, with a 10% difference between drug and placebo generally regarded as sufficient to establish antidepressant treatment advantage.18 Despite robust findings across efficacy measures in favor of levomilnacipran SR, response rates were lower than expected. MADRS response (≥ 50% decrease from baseline) was statistically different from placebo and exceeded the 10% threshold for clinical relevance for levomilnacipran SR 120 mg only.
Similar remission rates among levomilnacipran SR groups (21%–22%) and placebo (19%) may be due to limitations in the study design. The 8-week timeframe of an acute trial such as this is frequently not long enough to establish remission, especially in patients with high baseline MADRS scores (mean MADRS score in this study was 36). Additionally, the resulting number of patients achieving remission is likely
to be too small to detect between-treatment differences.18
Since MDD is a heterogeneous disorder, varying clinical outcomes may be the result of differences in patient populations. The Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study, the largest and longest study ever conducted in depression, was designed to systematically evaluate the effectiveness of depression treatments in generalizable samples in primary and specialty care settings. In STAR*D, only modest rates of remission (27.5% [HDRS score ≤ 7]) after up to 14 weeks of citalopram treatment were demonstrated, and, of patients achieving remission, 40% did so only after ≥ 8 weeks.19 Baseline characteristics associated with lower remission rates in STAR*D included high baseline depression severity and chronicity of illness,19 with the lowest rates and slower onset of remission seen in patients with chronic index depression episodes. Among remitted patients, both chronicity and recurrence were associated with higher risk of relapse in STAR*D.20 Of note, patients in the current study had similar baseline characteristics to those associated with the low remission rates seen in STAR*D, including high mean baseline depression severity, chronicity of illness, and recurrent depression (eg, in the current study, mean MADRS baseline score was 36, mean duration of symptoms was 11 years, and 76% of patients had recurrent depression).
No new safety concerns were revealed in the study. Levomilnacipran SR was generally well tolerated, even though more levomilnacipran SR patients than placebo patients discontinued because of AEs. A dose-response
relationship in tolerability was not supported, as the number of patients with AEs and discontinuation due to AEs was higher in the 80-mg group relative to the 40-mg and 120-mg groups.
AST and ALT levels were slightly increased at the end of double-blind treatment across all levomilnacipran SR doses relative to placebo. Increases were more pronounced in the levomilnacipran SR 80-mg dose group than in the 40-mg and 120-mg groups. The noted increases were largely due to 7 patients across the dose groups with postbaseline ALT and/or AST levels that met PCS criteria (≥ 3 × ULN). No AST or ALT increase was reported as an SAE or resulted in discontinuation from the study, and no patients met the criteria for Hy’s law.
Levomilnacipran SR treatment was not associated with weight gain, and mean changes in supine blood pressure were modest and similar across groups. Levomilnacipran SR was associated with mean increase in supine pulse rate, but this increase did not appear to be dose related. No clinically meaningful difference in QTcF between placebo and levomilnacipran SR was seen. Mean increases in QTcB interval, consistent with increases in heart rate, were observed
in levomilnacipran SR patients. Incidences of suicidal ideation and suicidal behavior were low and similar between treatment groups.
The evaluation of 3 fixed doses in this study allowed investigation of potential dose response, minimum effective dose, and maximum tolerable dose for levomilnacipran SR. Efficacy results indicated numerically greater improvement with higher doses on several measures. Functional improvement was demonstrated by statistically significant improvement in SDS total score for levomilnacipran SR 80 mg and 120 mg versus placebo. Conversely, the 120-mg dose did not show worse tolerability relative to the 80-mg dose. These findings support the use of higher dose levels as needed for efficacy, including dosing as high as 120 mg/d, with minimal impact on tolerability in patients with severe depression.
The lack of an active comparator arm limits the ability to compare these results with other antidepressants. Additionally, generalizability is also limited by inclusion and exclusion criteria.
Levomilnacipran is the more active enantiomer of milnacipran, an SNRI that is approved only for the treatment of fibromyalgia in the United States. On the basis of double-blind trials versus placebo, tricyclic antidepressants (TCAs), or SSRIs,21–26 twice-daily milnacipran has shown efficacy in major depressive episodes and is approved for the treatment of depression in many countries outside the United States. Milnacipran studies in depression were conducted outside the United States a decade ago; as such, no valid comparison between levomilnacipran SR and milnacipran data can be made.
Levomilnacipran SR differs from other SNRIs in its relative selectivity for norepinephrine versus serotonin reuptake inhibition. Dual blockade of serotonin and norepinephrine reuptake by SNRIs is similar to the mechanism of action of many older TCAs. However, TCAs also have multiple additional pharmacologic properties that result in tolerability problems and harmful adverse effects; without these additional interactions, SNRIs offer efficacy with a lower AE burden and better safety compared with TCAs.27 Additionally, it is suggested that antidepressants with a prominent noradrenergic component, such as levomilnacipran SR, may be particularly effective in treating the noradrenergic symptom cluster in depression (eg, functional impairment, decreased concentration, lassitude, mental and physical slowing, decreased self-care).2 8
When considering optimal antidepressant treatment, patients often describe the importance of both symptom
resolution and normalization of functioning.29 Levomilnacipran SR demonstrated efficacy and generally good tolerability in this clinical trial of patients with MDD. Consistent with what patients identify as the important components of recovery, levomilnacipran SR significantly improved both depressive symptoms and functional impairment in patients with MDD.
Drug names: citalopram (Celexa and others), duloxetine (Cymbalta), eszopiclone (Lunesta), milnacipran (Savella), venlafaxine (Effexor and others), zaleplon (Sonata and others), zolpidem (Ambien, Edluar, and others).
Author affiliations: Department of Psychiatry and Behavioral Sciences, Albert Einstein College of Medicine, Montefiore Medical Center, Bronx, New York (Dr Asnis); and Clinical Development (Drs Bose and Greenberg and Mr Gommoll) and Biostatistics (Dr Chen), Forest Research Institute, Jersey City, New Jersey.
Potential conflicts of interest: Dr Asnis has received grants from Forest, Shire, and Takeda and is a consultant for Bristol-Myers Squibb. Drs Bose, Chen, and Greenberg and Mr Gommoll are employees of Forest Research Institute.
Funding/support: Supported by funding from Forest Laboratories, Inc
(New York, New York). Forest Laboratories, Inc was involved in the study design; the collection (via contracted clinical investigator sites), analysis,
and interpretation of data; and the decision to present these results.
Previous presentations: American College of Neuropsychopharmacology, December 4–8, 2011, Waikoloa, Hawaii ▪ American Psychiatric Association, May 5–9, 2012, Philadelphia, Pennsylvania ▪ New Clinical Drug Evaluation Unit, May 29–June 1, 2012, Phoenix, Arizona ▪ and Collegium Internationale Neuro-Psychopharmacologicum, June 3–7, 2012, Stockholm, Sweden.
Acknowledgments: Writing assistance and editorial support for the preparation of this manuscript was provided by Adam Ruth, PhD; Paul Ferguson, MS; and Carol Dyer, MS, of Prescott Medical Communications Group, Chicago, Illinois, contractors of Forest Research Institute.
REFERENCES
1. Israel JA. Remission in depression: definition and initial treatment approaches. J Psychopharmacol. 2006;20(suppl):5–10. doi:10.1177/1359786806064306 PubMed
2. Depoortère R, Auclair A, Assié M-B, et al. In vivo characterization of levomilnacipran, a balanced serotonin norepinephrine reuptake inhibitor. Poster presented at: 66th Annual Meeting of the Society of Biological Psychiatry; May 12–14, 2011; San Francisco, CA.
3. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. Washington, DC:
American Psychiatric Association; 2000.
4. Sheehan DV, Lecrubier Y, Sheehan KH, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation
of a structured diagnostic psychiatric interview for DSM-IV and ICD-10.
J Clin Psychiatry. 1998;59(suppl 20):22–33, quiz 34–57. PubMed
5. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134(4):382–389. doi:10.1192/bjp.134.4.382 PubMed
6. Posner K, Brown GK, Stanley B, et al. The Columbia–Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults.
Am J Psychiatry. 2011;168(12):1266–1277. doi:10.1176/appi.ajp.2011.10111704 PubMed
7. Sheehan DV, Harnett-Sheehan K, Raj BA. The measurement of disability. Int Clin Psychopharmacol. 1996;11(suppl 3):89–95. doi:10.1097/00004850-199606003-00015 PubMed
8. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23(1):56–62. doi:10.1136/jnnp.23.1.56 PubMed
9. Guy W. The Clinician Global Severity and Impression Scales. In: ECDEU Assessment Manual for Psychopharmacology. US Department of Health, Education and Welfare publication (ADM) 76-338. Rockville, MD: National Institute of Mental Health; 1976; 218–222.
10. Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9(7):811–818. doi:10.1002/sim.4780090710 PubMed
11. Kenward MG, Molenberghs G, Thijs H. Pattern-mixture models with proper time dependence. Biometrika. 2003;90(1):53–71. doi:10.1093/biomet/90.1.53
12. Johnson WD, May WL. Combining 2 × 2 tables that contain structural zeros. Stat Med. 1995;14(17):1901–1911. doi:10.1002/sim.4780141706 PubMed
13. Mehta CR, Patel NR, Tsiatis AA. Exact significance testing to establish treatment equivalence with ordered categorical data. Biometrics. 1984;
40(3):819–825. doi:10.2307/2530927 PubMed
14. Nemeroff CB. The burden of severe depression: a review of diagnostic challenges and treatment alternatives. J Psychiatr Res. 2007;41(3–4):
189–206. doi:10.1016/j.jpsychires.2006.05.008 PubMed
15. Watkins PB, Seligman PJ, Pears JS, et al. Using controlled clinical trials to learn more about acute drug-induced liver injury. Hepatology. 2008;48(5):
1680–1689. doi:10.1002/hep.22633 PubMed
16. Sheehan KH, Sheehan DV. Assessing treatment effects in clinical trials with the discan metric of the Sheehan Disability Scale. Int Clin Psychopharmacol. 2008;23(2):70–83. doi:10.1097/YIC.0b013e3282f2b4d6 PubMed
17. Trivedi MH, Corey-Lisle PK, Guo Z, et al. Remission, response without remission, and nonresponse in major depressive disorder: impact on functioning. Int Clin Psychopharmacol. 2009;24(3):133–138. doi:10.1097/YIC.0b013e3283277614 PubMed
18. Montgomery SA, Möller HJ. Is the significant superiority of escitalopram compared with other antidepressants clinically relevant? Int Clin Psychopharmacol. 2009;24(3):111–118. doi:10.1097/YIC.0b013e32832a8eb2 PubMed
19. Trivedi MH, Rush AJ, Wisniewski SR, et al; STAR*D Study Team. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice.
Am J Psychiatry. 2006;163(1):28–40. doi:10.1176/appi.ajp.163.1.28 PubMed
20. Rush AJ, Wisniewski SR, Zisook S, et al. Is prior course of illness relevant
to acute or longer-term outcomes in depressed out-patients? a STAR*D report. Psychol Med. 2012;42(6):1131–1149. doi:10.1017/S0033291711002170 PubMed
21. Guelfi JD, Ansseau M, Corruble E, et al. A double-blind comparison of the efficacy and safety of milnacipran and fluoxetine in depressed inpatients. Int Clin Psychopharmacol. 1998;13(3):121–128. doi:10.1097/00004850-199805000-00005 PubMed
22. Kasper S, Pletan Y, Solles A, et al. Comparative studies with milnacipran and tricyclic antidepressants in the treatment of patients with major depression: a summary of clinical trial results. Int Clin Psychopharmacol. 1996;11(suppl 4):35–39. doi:10.1097/00004850-199609004-00005 PubMed
23. Lecrubier Y, Pletan Y, Solles A, et al. Clinical efficacy of milnacipran: placebo-controlled trials. Int Clin Psychopharmacol. 1996;11(suppl 4):
29–33. doi:10.1097/00004850-199609004-00004 PubMed
24. Lopez-Ibor J, Guelfi JD, Pletan Y, et al. Milnacipran and selective serotonin reuptake inhibitors in major depression. Int Clin Psychopharmacol. 1996;
11(suppl 4):41–46. doi:10.1097/00004850-199609004-00006 PubMed
25. Montgomery SA, Prost JF, Solles A, et al. Efficacy and tolerability of milnacipran: an overview. Int Clin Psychopharmacol. 1996;11(suppl 4):
47–51. doi:10.1097/00004850-199609004-00007 PubMed
26. Van Amerongen AP, Ferrey G, Tournoux A. A randomised, double-blind comparison of milnacipran and imipramine in the treatment of depression. J Affect Disord. 2002;72(1):21–31. doi:10.1016/S0165-0327(01)00422-0 PubMed
27. Nutt DJ. Relationship of neurotransmitters to the symptoms of major depressive disorder. J Clin Psychiatry. 2008;69 (suppl E1):4–7. PubMed
28. Kasper S, Meshkat D, Kutzelnigg A. Improvement of the noradrenergic symptom cluster following treatment with milnacipran. Neuropsychiatr
Dis Treat. 2011;7(suppl 1):21–27. PubMed
29. Zimmerman M, McGlinchey JB, Posternak MA, et al. How should remission from depression be defined? the depressed patient’s perspective. Am J Psychiatry. 2006;163(1):148–150. doi:10.1176/appi.ajp.163.1.148 PubMed
This PDF is free for all visitors!
![]() Save
Save
![]() Share
Share
![]() Cite
Cite