Introduction: Vilazodone is a potent serotonin (5-HT) reuptake inhibitor and 5-HT1A receptor partial agonist approved by the US Food and Drug Administration for the treatment of major depressive disorder (MDD) in adults. This study evaluated the efficacy and tolerability of vilazodone in the treatment of MDD.
Method: This 8-week, randomized (1:1), double-blind, placebo-controlled, parallel-group, fixed-dose study conducted from January 2012 to February 2013 compared vilazodone 40 mg/d with placebo in outpatients with DSM-IV-TR-diagnosed MDD. The primary efficacy measure was Montgomery-Asberg Depression Rating Scale (MADRS) total score change from baseline to week 8 analyzed by a mixed-effects model for repeated measures on the intent-to-treat population (placebo = 252, vilazodone = 253). Secondary efficacy outcomes were Clinical Global Impressions-Severity of Illness (CGI-S) Scale score change from baseline and MADRS sustained response rate (total score ≤ 12 for at least the last 2 consecutive double-blind visits).
Results: Approximately 83% of patients completed the study. Least squares mean differences (95% CI) were statistically significant for vilazodone versus placebo on MADRS (−5.117 [−6.886 to −3.347], P < .00001) and CGI-S (−0.622 [−0.845 to −0.399], P < .00001) change from baseline; statistically significant improvements versus placebo occurred at week 2 and persisted for the study duration. The MADRS sustained response rate was 17% for placebo and 27% for vilazodone (P < .01). Patients taking vilazodone versus placebo had higher rates of diarrhea and nausea; most incidences were mild in severity. Weight increase and sexual dysfunction adverse events were low in both groups.
Conclusions: A large and significant treatment effect on the MADRS and statistically significant improvement on the CGI-S demonstrated meaningful depressive symptom improvements. Vilazodone was generally well tolerated.
Trial Registration: ClinicalTrials.gov identifier: NCT01473394
Efficacy and Safety of Vilazodone in Major Depressive Disorder:
A Randomized, Double-Blind, Placebo-Controlled Trial
ABSTRACT
Introduction: Vilazodone is a potent serotonin (5-HT) reuptake inhibitor and 5-HT1A receptor partial agonist approved by the US Food and Drug Administration for the treatment of major depressive disorder (MDD) in adults. This study evaluated the efficacy and tolerability of vilazodone in the treatment of MDD.
Method: This 8-week, randomized (1:1), double-blind, placebo-controlled, parallel-group, fixed-dose study conducted from January 2012 to February 2013 compared vilazodone 40 mg/d with placebo in outpatients with DSM-IV-TR–diagnosed MDD. The primary efficacy measure was Montgomery-Asberg Depression Rating Scale (MADRS) total score change from baseline to week 8 analyzed by a mixed-effects model for repeated measures on the intent-to-treat population (placebo = 252, vilazodone = 253). Secondary efficacy outcomes were Clinical Global Impressions–Severity of Illness (CGI-S) Scale score change from baseline and MADRS sustained response rate (total score ≤ 12 for at least the last 2 consecutive double-blind visits).
Results: Approximately 83% of patients completed the study. Least squares mean differences (95% CI) were statistically significant for vilazodone versus placebo on MADRS (−5.117 [−6.886 to −3.347], P < .00001) and CGI-S (−0.622 [−0.845 to −0.399], P < .00001) change from baseline; statistically significant improvements versus placebo occurred at week 2 and persisted for the study duration. The MADRS sustained response rate was 17% for placebo and 27% for vilazodone (P < .01). Patients taking vilazodone versus placebo had higher rates of diarrhea and nausea; most incidences were mild in severity. Weight increase and sexual dysfunction adverse events were low in both groups.
Conclusions: A large and significant treatment effect on the MADRS and statistically significant improvement on the CGI-S demonstrated meaningful depressive symptom improvements. Vilazodone was generally well tolerated.
Trial Registration: ClinicalTrials.gov identifier: NCT01473394
J Clin Psychiatry 2014;75(11):e1291–e1298
© Copyright 2014 Physicians Postgraduate Press, Inc.
Submitted: January 9, 2014; accepted April 4, 2014 (doi:10.4088/JCP.14m08992).
Corresponding author: Harry A. Croft, MD, 7434 Louis Pasteur,
Ste 101, San Antonio, TX 78229 ([email protected]).
Serotonin (5-HT) reuptake inhibitor antidepressants, including selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs), give clinicians numerous pharmacotherapy options for treating major depressive disorder (MDD). Not all antidepressants are effective in all patients; choice of antidepressant may be influenced by factors including efficacy, clinical characteristics, medical and psychiatric comorbidities, and the desire to minimize specific adverse events such as weight gain and sexual dysfunction.1
While all serotonin reuptake inhibitors enhance overall serotonergic transmission to achieve antidepressant activity, pharmacology differences influence both efficacy and tolerability profiles. Faster expected time to onset, efficacy in comorbid conditions (such as anxiety), and persistence of treatment response are characteristics that may be associated with specific mechanisms of action.2–4 Since many patients do not achieve remission following initial treatment5 and most patients have residual symptoms, which are associated with lower quality of life and greater risk for relapse,6 antidepressant options are essential.
Vilazodone is a SSRI and 5-HT1A receptor partial agonist approved for the treatment of MDD in adults. Although the net effect of 5-HT1A partial agonism on serotonergic transmission is not yet known, some evidence suggests that activating 5-HT1A receptors may enhance antidepressant efficacy by improving time to onset of action and augmenting anxiolytic effects.7–9 Vilazodone efficacy was established in two 8-week, double-blind, placebo-controlled phase III trials (ClinicalTrials.gov identifiers: NCT00285376 and NCT00683592).10,11 In both studies, vilazodone showed significantly greater improvement relative to placebo on the primary efficacy outcome, Montgomery-Asberg Depression Rating Scale (MADRS) total score mean change from baseline to week 8. Safety and tolerability findings were supported in a 1-year, open-label trial of vilazodone 40 mg/d (ClinicalTrials.gov identifier: NCT00644358).12 Vilazodone was generally well tolerated in all trials; common adverse events, including diarrhea, nausea, and insomnia, were generally transient in nature and mild to moderate in severity.13 The objective of the current study (ClinicalTrials.gov identifier: NCT01473394) was to further characterize the efficacy, safety, and tolerability of vilazodone 40 mg/d versus placebo for the treatment of MDD; a novel prospectively defined secondary endpoint (MADRS sustained response) was also assessed in this trial to evaluate treatment response that persisted beyond a single time point.
METHOD
This study was conducted at 14 US study centers between January 2012 and February 2013 in full compliance with US Food and Drug Administration (FDA) guidelines for Good Clinical Practice and the ethical principles of the Declaration of Helsinki. The protocol was approved by institutional review boards, and all patients provided written informed consent.
Study Design
This was a multicenter, randomized, double-blind, placebo-controlled, parallel-group, fixed-dose study comparing vilazodone with placebo in outpatients with MDD. The study consisted of a 1- to 4-week no-drug screening period, 8-week double-blind treatment, and a 1-week double-blind taper period (vilazodone 20 mg/d for 4 days and 10 mg/d for 3 days). Eligible patients were randomized (1:1) to receive placebo or vilazodone 40 mg/d. Vilazodone was initiated at 10 mg/d for week 1; dosage was increased to 20 mg/d for week 2 and to 40 mg/d for weeks 3–8. All study drug was taken once daily with food.
Patients were randomized by computer-generated number to identically appearing treatment. Investigators and patients were blinded to study drug allocation; the blind was maintained via a secured randomization code list and broken only in case of emergency. Unblinding disqualified a patient from further participation. All efficacy assessments were conducted in person at each investigative center by experienced clinicians who met the training requirements and qualification standards that were established by the sponsor and the rater training vendor. The raters were assessed annually on their ability to score the relevant primary efficacy scale compared with acceptable scores established by a combination of expert opinion scores, group modal scores, and clinical analysis.
Inclusion Criteria
Male or female outpatients (18–70 years, inclusive) who met Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR)14 criteria for MDD, with an ongoing major depressive episode ≥ 8 weeks’ and ≤ 12 months’ duration and MADRS15 total score ≥ 26 were included. Patients had normal (or abnormal results that were judged to be not clinically significant) physical examination, clinical laboratory, and electrocardiogram (ECG) findings. Body mass index between 18 and 40 kg/m2, inclusive, was required. All women of childbearing potential were required to have a negative β-hCG pregnancy test and use a reliable method of contraception.
Exclusion Criteria
Typical exclusion criteria for antidepressant clinical trials were applied. Psychiatric exclusions included a DSM-IV-TR–defined Axis I disorder other than MDD within 6 months of study (secondary comorbid generalized anxiety disorder, social anxiety disorder, and/or specific phobias were allowed), history of other DSM diagnoses (eg, bipolar, obsessive-compulsive, psychotic, or cognitive disorder), or substance abuse/dependence within 6 months of study. Patients with suicide risk were excluded (ie, past year attempt, MADRS suicidal thoughts item score ≥ 5, Columbia-Suicide Severity Rating Scale [C-SSRS]16 findings).
Treatment-related exclusions included nonresponse to ≥ 2 antidepressants, intolerance/hypersensitivity to vilazodone/SNRIs/SSRIs, or treatment with prohibited medications (including any psychotropic drug, any drug with psychotropic activity, any drug with a potentially psychotropic component, or any medication that is a strong cytochrome P450 3A4 or 2C19 inhibitor or inducer). Eszopiclone, zopiclone, zaleplon, zolpidem, or zolpidem extended release could be used for insomnia. Medical conditions that could interfere with study conduct, confound interpretation of results, or endanger patient well-being were exclusionary.
Efficacy and Safety Assessments
The primary efficacy measure was the MADRS (assessed at week −1 [screening], baseline [week 0], weeks 1, 2, 4, 6, 8).
Clinical Global Impressions–Severity of Illness (CGI-S) Scale,17 a secondary outcome, was assessed on the same schedule. Additional efficacy measures included CGI-Improvement (CGI-I) Scale17 (weeks 1, 2, 4, 6, 8) and Hamilton Anxiety Rating Scale (HAM-A)18 (weeks 0, 2, 4, 6, 8). Safety was evaluated by adverse event reports, physical examination, clinical laboratory and vital sign measures, ECGs, and C-SSRS findings (all visits).
Statistical Analyses
Safety analyses were based on the safety population (all randomized patients who received ≥ 1 dose of double-blind study drug); efficacy analyses were based on the intent-to-treat population (patients in the safety population who had baseline and ≥ 1 postbaseline MADRS assessments).
The prespecified primary efficacy outcome was MADRS total score change from baseline to week 8; the primary analysis was a mixed-effects model for repeated measures (MMRM) with treatment group, study center, visit, and treatment group by visit interaction as fixed effects and the baseline value and baseline value by visit interaction as covariates. An unstructured covariance matrix was used to model the covariance of within-patient scores. The Kenward-Roger approximation19 was used to estimate the denominator degrees of freedom. Two sensitivity analyses were performed on the primary outcome: a pattern-mixture model (PMM) based on non–future-dependent missing value restrictions20 to the possible violation of the missing-at-random assumption and an analysis of covariance (ANCOVA) based on the last observation carried forward (LOCF) approach.
The prospectively defined secondary efficacy parameters were change from baseline to week 8 in CGI-S score and MADRS sustained response rate. The CGI-S score was analyzed using an MMRM approach similar to the primary measure; sustained response was analyzed using the Cochran-Mantel-Haenszel test controlling for study center. For this study, sustained response was defined as MADRS total score ≤ 12 for at least the last 2 consecutive double-blind visits. This criterion was chosen in consultation with the FDA and was agreed to as a key secondary outcome to show nontransient improvement. Sensitivity analyses performed on the secondary outcomes comprised a PMM approach for sustained response and an ANCOVA based on the LOCF approach for CGI-S score change from baseline.
Change from baseline to endpoint in HAM-A scores and CGI-I score at endpoint were analyzed using an MMRM model similar to the primary analyses. MADRS (≥ 50% improvement from total score baseline) and CGI-I (score ≤ 2) response rates were additional efficacy outcomes analyzed using a generalized linear mixed model. Demographic and baseline characteristics were tested using a 2-way analysis of variance with treatment group and study center as factors for continuous variables and the Cochran-Mantel-Haenszel test, controlling for study center, for categorical variables.
Hierarchical testing was applied to control type I error at the .05 significance level. The CGI-S outcome was only tested inferentially if the primary efficacy analysis (MADRS) was positive (P < .05). The sustained response efficacy outcome was not inferentially tested unless the CGI-S analysis was positive. Statistical testing on additional endpoints was performed without adjustment for multiple comparisons; reported P values are nominal. Effect sizes for change from baseline in MADRS, CGI-S, and HAM-A scores and for the CGI-I score were calculated using Cohen d. Descriptive statistics were used for all safety parameters.
RESULTS
Patient Disposition and Demographic Characteristics
Study populations, patient disposition, and demographic characteristics are summarized in Table 1. There were no significant between-group differences in discontinuation rates, overall or for any individual reason.
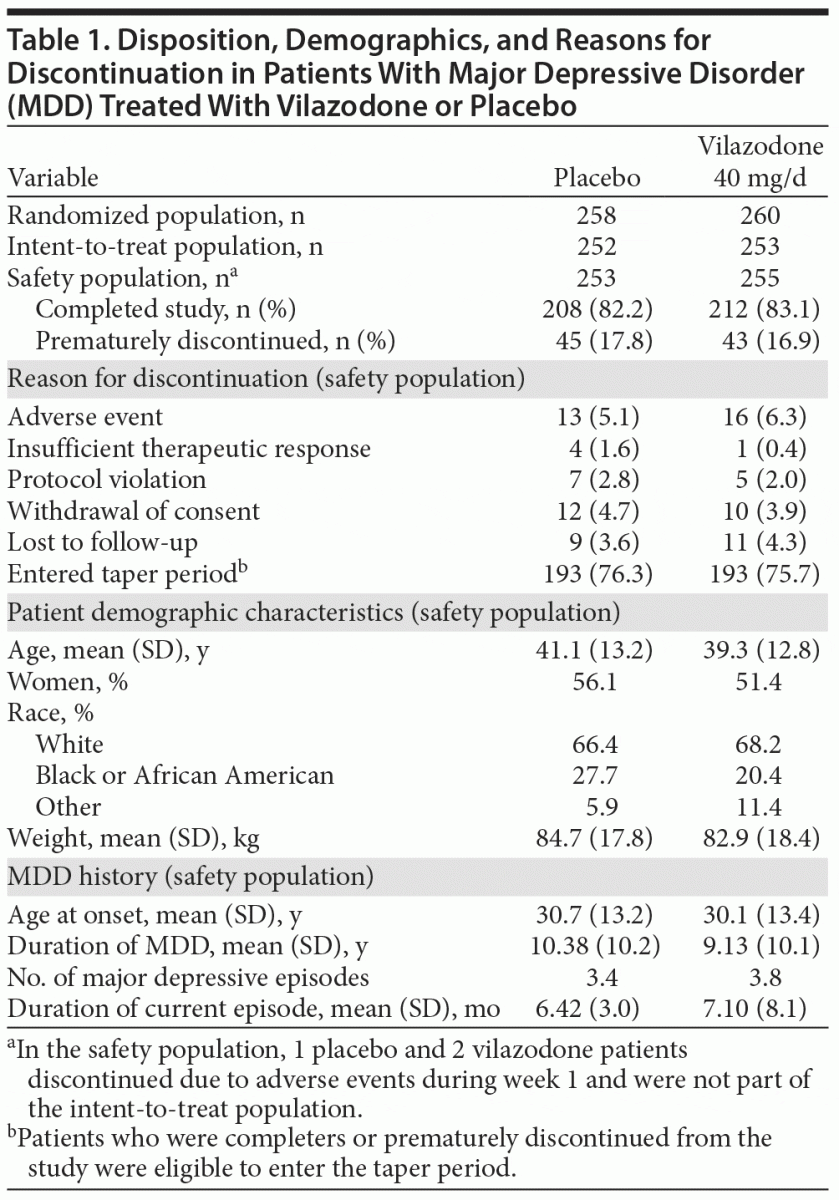
There were no significant between-group differences in any demographic or physical characteristic (Table 1); 71% of patients were 20–49 years of age, 18% were 50–59 years, and 9% were ≥ 60 years. Mean MADRS baseline total scores were ~ 30 in both groups, indicating a patient population with at least moderate MDD symptoms.
Efficacy Outcomes
On MADRS total score change from baseline to week 8 (primary efficacy), statistically significant reductions that were consistent with greater symptom improvement were seen for vilazodone- versus placebo-treated patients (least squares mean difference [LSMD] = −5.117, P < .00001, effect size = 0.54); LOCF and PMM (data not shown) sensitivity analyses supported the primary results (Table 2). Statistically significant differences in treatment effect for vilazodone were apparent at week 2 and increased over time throughout double-blind treatment (Figure 1A).
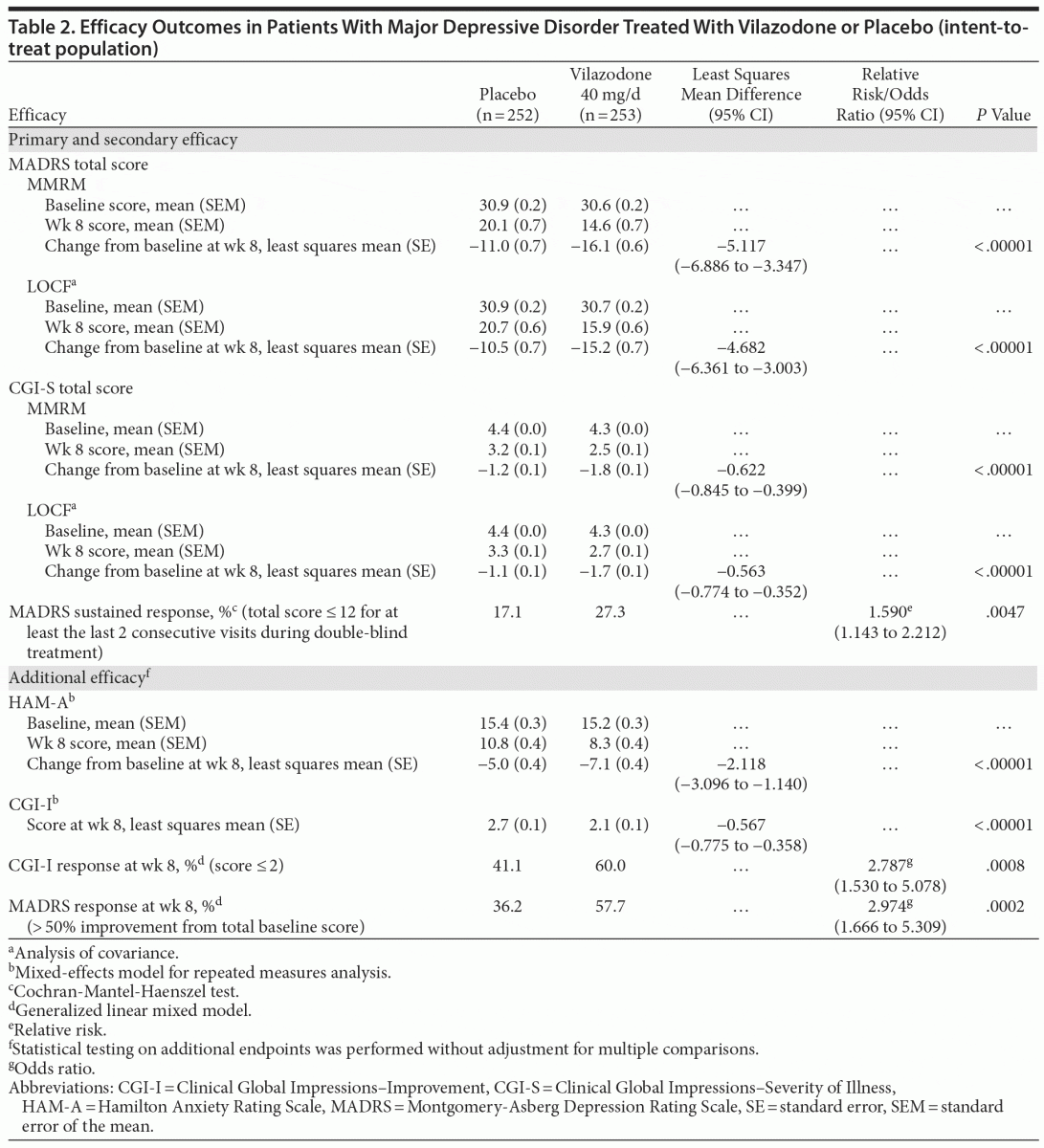
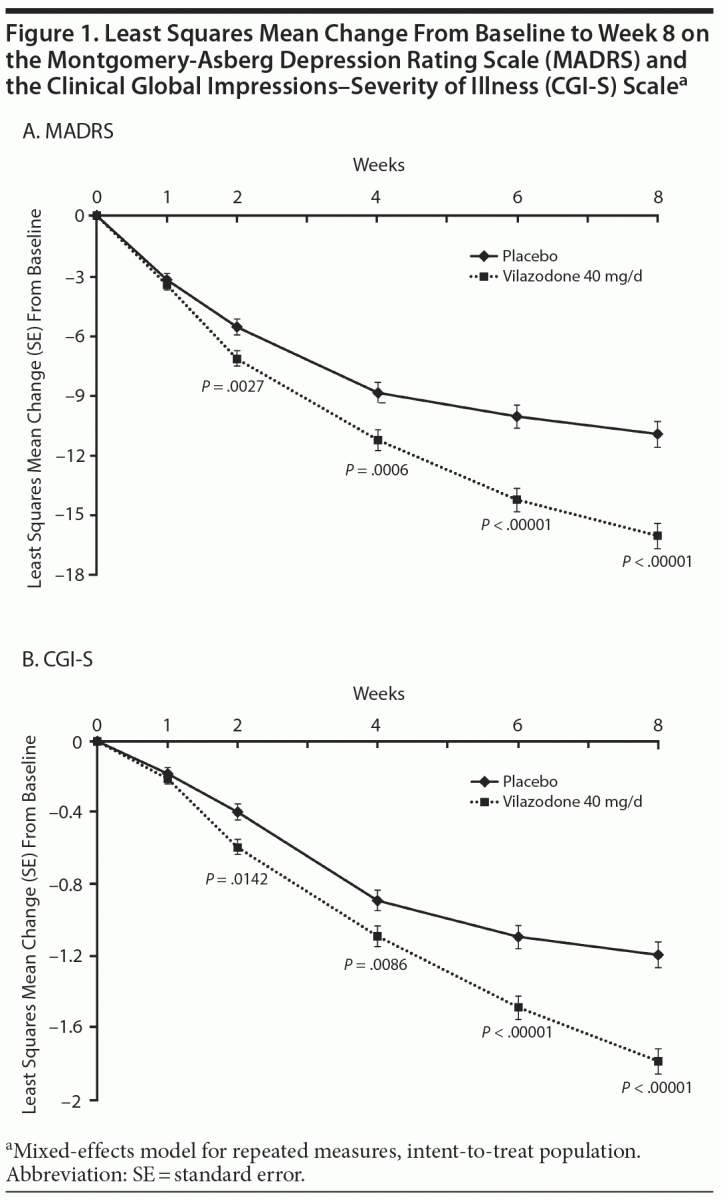
Statistically significant differences were also seen for vilazodone- versus placebo-treated patients on the secondary outcome measures. Decrease from baseline to week 8 in CGI-S score was statistically greater for vilazodone versus placebo (LSMD = −0.622, P < .00001, effect size = 0.50); LOCF sensitivity analysis supported MMRM results (Table 2). A statistically significant difference in favor of vilazodone was seen at week 2; the treatment effect increased over time to week 8 (Figure 1B). The difference in the rate of MADRS sustained response was also statistically significant in favor of vilazodone (27%) versus placebo (17%, P = .0047) (Table 2).
At week 8, statistically significant differences for vilazodone versus placebo were observed on all additional efficacy parameters (Table 2); effect sizes for change from baseline in HAM-A scores and CGI-I score at endpoint were 0.39 and 0.43, respectively. Additionally, once the difference between groups reached statistical significance, it was maintained until the end of treatment (HAM-A: week 4 [P = .0080], week 6 [P = .0006], week 8 [P < .00001]; MADRS response rate: week 6 [P = .0005], week 8 [P = .0002]; CGI-I response rate: week 6 [P = .0092], week 8 [P = .0008]).
Safety and Tolerability
Extent of exposure. The median duration of treatment was 56 days for both groups. During double-blind treatment, the mean (SD) total daily vilazodone dose was 31.4 (5.4) mg; the final mean (SD) daily dose was 38.5 (5.9) mg.
Adverse events. A summary of adverse events is presented in Table 3; adverse events that occurred in > 5% of vilazodone patients and at 2 times the rate of placebo were diarrhea, nausea, dizziness, and insomnia. During double-blind treatment, most treatment-emergent adverse events were considered by the investigator to be mild or moderate in severity (placebo = 97%, vilazodone = 99%) and possibly related to the study drug (placebo = 64%, vilazodone = 79%).
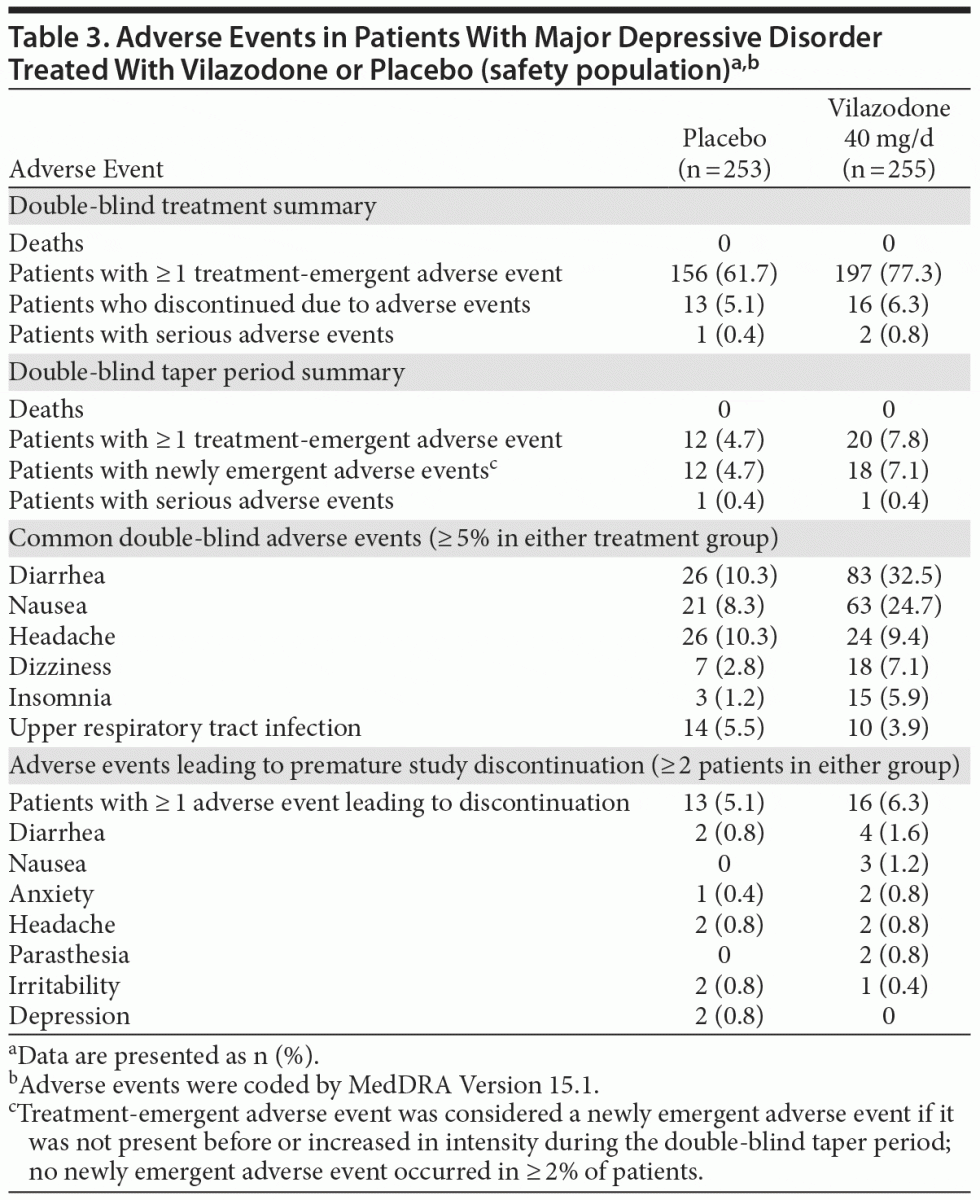
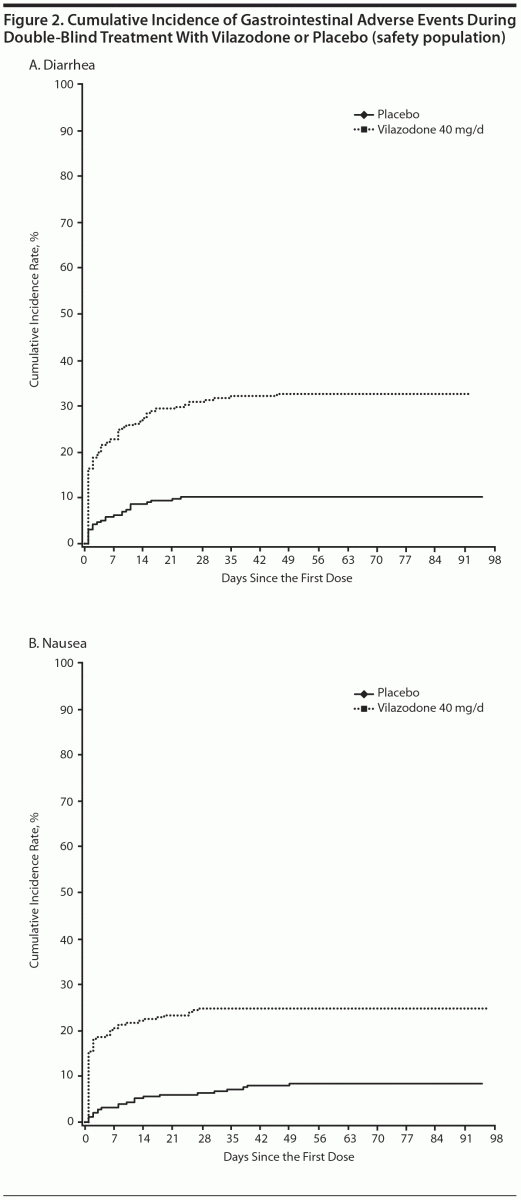
Diarrhea and nausea occurred early in the course of treatment (day 1) and were transient in nature (Figures 2A and B). In vilazodone patients who reported diarrhea and nausea, 75% and 79% of cases, respectively, were considered mild in severity; diarrhea and nausea led to the discontinuation of 4 (1.6%) and 3 (1.2%) patients, respectively. Although the incidence of sexual dysfunction adverse events was relatively low in both groups, more male patients treated with vilazodone versus placebo reported erectile dysfunction (6 [4.8% vs 0.9%]) and delayed ejaculation (3 [2.4% vs 0]).
During double-blind treatment, serious adverse events were reported in 1 placebo patient (breast cancer [discontinued study]) and 2 vilazodone patients (noncardiac chest pain and suicidal ideation [discontinued study] in 1 patient each); no double-blind serious adverse event was considered related to study drug.
During the double-blind taper period, serious adverse events were reported in 1 placebo patient (intentional overdose and suicide attempt; considered related to study drug) and 1 vilazodone patient (myocardial infarction; considered not related to study drug).
Clinical laboratory, vital sign, electrocardiogram evaluation. Mean changes from baseline in liver enzymes, most laboratory parameters, and vital signs (including blood pressure, pulse rate, and body weight) were small and similar between groups. No patient met Hy’s law criteria (alanine aminotransferase or aspartate aminotransferase elevation ≥ 3 × ULN [upper limit of normal], total bilirubin elevation > 2 × ULN, and alkaline phosphatase < 2 × ULN).21 Potentially clinically significant values were generally low and similar for placebo and vilazodone. The most frequent potentially clinically significant laboratory abnormality was elevated creatine kinase (placebo, 5%; vilazodone, 10%).
Mean weight gain was 0.10 kg (0.22 lb) for placebo patients and 0.37 kg (0.82 lb) for vilazodone patients; potentially clinically significant weight increase (≥ 7% from baseline) was low for both placebo (0.4%) and vilazodone (1%) groups.
Orthostatic hypotension (≥ 20 mm Hg reduction in systolic blood pressure or ≥ 10 mm Hg reduction in diastolic blood pressure while changing from supine to standing position) was reported in 15 (6%) placebo- and 13 (5%) vilazodone-treated patients; no consistent orthostatic hypotension adverse event pattern was noted. No patient had a QTcB or QTcF (QT interval corrected for heart rate using Bazett or Fridericia formula, respectively) value > 500 msec or a clinically significant ECG.
C-SSRS suicidality and suicide-related adverse events. The incidence of C-SSRS–rated suicidal ideation was similar between groups (placebo, 21%; vilazodone, 19%). There were no attempted suicides, no interrupted suicide attempts, and 1 aborted suicide attempt (placebo patient) during the double-blind period. Suicidality-related serious adverse events were reported by 2 vilazodone patients (suicidal ideation on day 1 of double-blind treatment in 1 patient and suicide attempt by intentional overdose with marketed vilazodone [Viibryd] 3 days after the last dose of double-blind taper placebo in 1 patient); both patients discontinued treatment and recovered.
DISCUSSION
In this randomized, placebo-controlled, double-blind study, greater improvement in depressive symptoms for patients treated with vilazodone 40 mg/d compared with placebo was demonstrated by statistically significant differences in mean MADRS total score change from baseline to week 8. The magnitude of MADRS improvement (LSMD = −5.117, P < .00001) suggests that vilazodone effectively treated the symptoms of depression. Short-term antidepressant efficacy in clinical trials is typically measured by improvement on a standard depression symptoms rating scale; a mean 2-point difference versus placebo on the MADRS total score is frequently used as a threshold to indicate the clinical relevance of active treatment.22 As such, the greater than 5-point LSMD in favor of vilazodone on the primary efficacy measure in this study exceeds what is considered a clinically relevant antidepressant effect.
Improvements on secondary and additional efficacy measures also suggested that vilazodone was associated with improvements across diverse outcomes including reduced disease severity, clinical global improvement, and persistent treatment response. Vilazodone-treated patients also showed significant but modest improvement in anxiety symptoms relative to placebo patients. Limited improvement in anxiety was probably due to the low baseline HAM-A scores (mean and median baseline scores were approximately 15), suggesting many patients had only mild anxiety symptoms at baseline.
Given the debilitating nature of depressive symptoms, early improvement and persistent efficacy are clinically relevant from both the clinical and patient perspective. In this study, statistically significant separation for vilazodone versus placebo on MADRS total score reduction occurred as early as week 2 and was sustained for the remainder of treatment, suggesting that the observed improvement was not transient in nature. Additionally, early improvement was observed and maintained throughout treatment on the CGI-S, CGI-I, and HAM-A.
The ability to demonstrate enduring change is limited by the short duration of antidepressant clinical trials. The concept of sustained response was developed in consultation with the FDA as a way to demonstrate when treatment benefit is maintained beyond a single time point in a short-term study. The criterion used to define sustained response in this study (MADRS score ≤ 12 for at least the last 2 consecutive visits) is relatively stringent, requiring a MADRS score that indicates a low level of depression symptoms.23 In the current study, the rate of MADRS sustained response was statistically significant (P < .01) in favor of vilazodone- (27%) versus placebo-treated (17%) patients, indicating that vilazodone patients achieved low levels of depressive symptoms and improvement was not transient.
Vilazodone 40 mg/d was previously evaluated in two 8-week phase III clinical trials of similar design in adult patients with MDD.10,11 The LSMD (95% CI) for MADRS change from baseline for vilazodone versus placebo was −3.2 (−5.1 to −1.2) and −2.5 (−4.3 to −0.6) in the previous trials compared with −5.1 in the current trial. Response rates for vilazodone were also considerably higher in the current study (57.7%) relative to the previous studies (40.4% and 43.7%); in comparison, placebo response rates were only marginally higher in the current study (36.2%) versus the previous studies (28.1% and 30.3%).10,11 The LSMDs for vilazodone versus placebo for CGI-S, HAM-A, and CGI-I were also greater in this study compared with previous studies. Reasons for greater magnitude of effect seen in this study relative to earlier studies is unknown but may be due to differences in the underlying patient population and/or differences in design and conduct of the studies.
A clinician’s choice of antidepressant treatment is influenced by multiple factors including a patient’s clinical presentation and the tolerability profile of the medication.1 Approximately half of patients diagnosed with MDD also have clinically meaningful levels of anxiety.24,25 Mean weight gain of 6.8–10.8 kg (15.0–23.8 lb) has been reported during long-term SSRI therapy,26 and sexual dysfunction, already a common problem for patients with MDD,27 is reported in approximately 40% of patients treated with SSRIs and SNRIs.28 In a study by Hu et al,29 weight gain and sexual dysfunction, along with drowsiness, were considered by patients to be the 3 most frequently bothersome side effects associated with antidepressant treatment. Treating both depression and anxiety symptoms, as well as avoiding sexual dysfunction and weight gain adverse events, have been identified as important for clinicians who prescribe antidepressants.1
In the current study, early and statistically significant differences in treating symptoms of depression and associated anxiety were demonstrated for vilazodone versus placebo. Mean weight gain was small and similar for vilazodone and placebo, supporting results observed in the pivotal trials of vilazodone10,12; however, weight gain is often not detected in acute 8-week trials, and longer duration may be necessary to reliably detect changes. In a previous long-term open-label study,12 mean weight increase was 1.7 kg (3.7 lb) after 52 weeks of vilazodone treatment versus 0.37 kg (0.82 lb) in the current 8-week study.
The incidence of sexual dysfunction treatment-emergent adverse events was < 5% in both groups, which is comparable to previous vilazodone trials.10–12 Spontaneous reporting of sexual dysfunction adverse events may underestimate sexual dysfunction, and this study did not include a scale to measure sexual function, so the sexual function results must be interpreted accordingly. Results of previous vilazodone studies that used prospective sexual dysfunction measures have shown that sexual functioning was similar for vilazodone- and placebo-treated patients in spite of prominent baseline sexual dysfunction.10–12,30 Additionally, an 8-week trial may be too short of a time period to accurately assess sexual dysfunction. However, in a long-term 52-week open-label study of vilazodone, reports of sexual dysfunction treatment-emergent adverse events were similar to the 2 phase III 8-week trials.30
Gastrointestinal adverse event incidence in the current study is consistent with observations from the vilazodone registration studies and a 52-week open-label safety study10–12; as such, they were not unexpected here. Similar to prior studies, most instances of vilazodone-related diarrhea and nausea were mild or moderate in intensity, led to few premature discontinuations, occurred in the first few weeks of treatment, and were transient in nature.
Vilazodone was otherwise generally well tolerated in this study. Adverse events indicative of cardiovascular effects (eg, tachycardia, palpitations, chest pain, hypertension, orthostatic hypotension) occurred in < 2% of patients in either treatment group, and no notable between-group differences were observed for laboratory values, liver function tests, vital signs, or ECGs.
Limitations of the current study include short duration and inclusion/exclusion criteria that may restrict generalizability. The lack of an active comparator limits the ability to draw conclusions versus other antidepressants. No prospective measure was used to evaluate sexual dysfunction. Sustained response is not a consistently defined or validated measure in antidepressant clinical trials; as such, findings should be interpreted with discretion.
CONCLUSION
The large MADRS treatment effect (LSMD vs placebo = −5.117) achieved in this study is notable since the clinical objective of MDD treatment is depressive symptom resolution. Additionally, MADRS, CGI-S, and HAM-A improvements occurred early and were maintained for the duration of treatment. Common vilazodone-related gastrointestinal adverse events were generally mild and transient and did not interfere with treatment continuation for most patients. This study supports the efficacy, safety, and tolerability observed in the pivotal studies and reinforces the merits of vilazodone as an important treatment option for adults with MDD.
Drug names: eszopiclone (Lunesta), zaleplon (Sonata and others), vilazodone (Viibryd), zolpidem (Ambien, Edluar, and others).
Author affiliations: San Antonio Psychiatric Research Center, San Antonio, Texas (Dr Croft); Nathan Kline Institute, New York University School of Medicine, Orangeburg, New York (Dr Pomara); and Forest Research Institute, Jersey City, New Jersey (Drs Chen, Nunez, and Mathews and Mr Gommoll).
Potential conflicts of interest: Dr Croft has received research grants from AstraZeneca, Boehringer Ingelhelim, Bristol-Myers Squibb, Cephalon, Clinical Data, Elan, Forest, GlaxoSmithKline, Labopharm, Eli Lilly, Lundbeck, Merck, Otsuka, Pfizer, Sarnoff, Sepracor, Shire, Takeda, and Trimel; has served as a consultant to Boehringer Ingelhelim, Clinical Data, Forest, and GlaxoSmithKline; and has served as a speaker for AstraZeneca, Boehringer Ingelhelim, Eli Lilly, Forest, Pfizer, and Takeda. Drs Chen, Nunez, and Mathews and Mr Gommoll are employees of Forest Research Institute. Dr Pomara reports no conflict of interest related to the subject of this article.
Funding/support: Supported by funding from Forest Laboratories, Inc (New York, New York).
Role of the sponsor: Forest Laboratories, Inc, was involved in the study design, the collection of data (via contracted clinical investigator sites), the analysis and interpretation of data, and the decision to present these results.
Previous presentation: American College of Neuropsychopharmacology; December 8–12, 2013; Hollywood, Florida.
Acknowledgments: Writing assistance and editorial support for the preparation of this manuscript were provided by Adam Ruth, PhD, and Carol Dyer, MS, of Prescott Medical Communications Group, Chicago, Illinois, a contractor of Forest Research Institute. Dr Ruth and Ms Dyer report no other conflicts of interest related to the subject of this article.
REFERENCES
1. Zimmerman M, Posternak M, Friedman M, et al. Which factors influence psychiatrists’ selection of antidepressants? Am J Psychiatry. 2004;161(7):1285–1289. doi:10.1176/appi.ajp.161.7.1285 PubMed
2. Nutt DJ. Relationship of neurotransmitters to the symptoms of major depressive disorder. J Clin Psychiatry. 2008;69(suppl E1):4–7. PubMed
3. Taylor MJ. Rapid onset of true antidepressant action. Curr Psychiatry Rep. 2007;9(6):475–479. doi:10.1007/s11920-007-0064-0 PubMed
4. Taylor MJ, Freemantle N, Geddes JR, et al. Early onset of selective serotonin reuptake inhibitor antidepressant action: systematic review and meta-analysis. Arch Gen Psychiatry. 2006;63(11):1217–1223. doi:10.1001/archpsyc.63.11.1217 PubMed
5. Trivedi MH, Rush AJ, Wisniewski SR, et al; STAR*D Study Team. Evaluation of outcomes with citalopram for depression using measurement-based care in STAR*D: implications for clinical practice. Am J Psychiatry. 2006;163(1):28–40. doi:10.1176/appi.ajp.163.1.28 PubMed
6. Zajecka JM. Treating depression to remission. J Clin Psychiatry. 2003;64(suppl 15):7–12. PubMed
7. Albert PR, François BL. Modifying 5-HT1A receptor gene expression as a new target for antidepressant therapy. Front Neurosci. 2010;4:35. PubMed
8. Blier P, Ward NM. Is there a role for 5-HT1A agonists in the treatment of depression? Biol Psychiatry. 2003;53(3):193–203. doi:10.1016/S0006-3223(02)01643-8 PubMed
9. Portella MJ, de Diego-Adeliño J, Ballesteros J, et al. Can we really accelerate and enhance the selective serotonin reuptake inhibitor antidepressant effect? a randomized clinical trial and a meta-analysis of pindolol in nonresistant depression. J Clin Psychiatry. 2011;72(7):962–969. doi:10.4088/JCP.09m05827blu PubMed
10. Khan A, Cutler AJ, Kajdasz DK, et al. A randomized, double-blind, placebo-controlled, 8-week study of vilazodone, a serotonergic agent for the treatment of major depressive disorder. J Clin Psychiatry. 2011;72(4):441–447. doi:10.4088/JCP.10m06596 PubMed
11. Rickels K, Athanasiou M, Robinson DS, et al. Evidence for efficacy and tolerability of vilazodone in the treatment of major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(3):326–333. doi:10.4088/JCP.08m04637 PubMed
12. Robinson DS, Kajdasz DK, Gallipoli S, et al. A 1-year, open-label study assessing the safety and tolerability of vilazodone in patients with major depressive disorder. J Clin Psychopharmacol. 2011;31(5):643–646. PubMed
13. Liebowitz M, Croft HA, Kajdasz DK, et al. The safety and tolerability profile of vilazodone, a novel antidepressant for the treatment of major depressive disorder. Psychopharmacol Bull. 2011;44(3).
14. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
15. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134(4):382–389. doi:10.1192/bjp.134.4.382 PubMed
16. Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266–1277. doi:10.1176/appi.ajp.2011.10111704 PubMed
17. Guy W. The clinician global severity and impression scales. In: ECDEU Assessment Manual for Psychopharmacology. US Dept Health, Education, and Welfare publication (ADM) 76-338. Rockville, MD: National Institute of Mental Health; 1976;218–222.
18. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23(1):56–62. doi:10.1136/jnnp.23.1.56 PubMed
19. Kenward MG, Roger JH. Small sample inference for fixed effects from restricted maximum likelihood. Biometrics. 1997;53(3):983–997. doi:10.2307/2533558 PubMed
20. Kenward MG, Molenberghs G, Thijs H. Pattern-mixture models with proper time dependence. Biometrika. 2003;90(1):53–71. doi:10.1093/biomet/90.1.53
21. Watkins PB, Seligman PJ, Pears JS, et al. Using controlled clinical trials to learn more about acute drug-induced liver injury. Hepatology. 2008;48(5):1680–1689. doi:10.1002/hep.22633 PubMed
22. Khin NA, Chen YF, Yang Y, et al. Exploratory analyses of efficacy data from major depressive disorder trials submitted to the US Food and Drug Administration in support of new drug applications. J Clin Psychiatry. 2011;72(4):464–472. doi:10.4088/JCP.10m06191 PubMed
23. Montgomery SA, Möller HJ. Is the significant superiority of escitalopram compared with other antidepressants clinically relevant? Int Clin Psychopharmacol. 2009;24(3):111–118. doi:10.1097/YIC.0b013e32832a8eb2 PubMed
24. Rao S, Zisook S. Anxious depression: clinical features and treatment. Curr Psychiatry Rep. 2009;11(6):429–436. doi:10.1007/s11920-009-0065-2 PubMed
25. Wiethoff K, Bauer M, Baghai TC, et al. Prevalence and treatment outcome in anxious versus nonanxious depression: results from the German Algorithm Project. J Clin Psychiatry. 2010;71(8):1047–1054. doi:10.4088/JCP.09m05650blu PubMed
26. Ferguson JM. SSRI antidepressant medications: adverse effects and tolerability. Prim Care Companion J Clin Psychiatry. 2001;3(1):22–27. doi:10.4088/PCC.v03n0105 PubMed
27. Clayton AH, Montejo AL. Major depressive disorder, antidepressants, and sexual dysfunction. J Clin Psychiatry. 2006;67(suppl 6):33–37. PubMed
28. Clayton AH, Pradko JF, Croft HA, et al. Prevalence of sexual dysfunction among newer antidepressants. J Clin Psychiatry. 2002;63(4):357–366. doi:10.4088/JCP.v63n0414 PubMed
29. Hu XH, Bull SA, Hunkeler EM, et al. Incidence and duration of side effects and those rated as bothersome with selective serotonin reuptake inhibitor treatment for depression: patient report versus physician estimate. J Clin Psychiatry. 2004;65(7):959–965. doi:10.4088/JCP.v65n0712 PubMed
30. Clayton AH, Kennedy SH, Edwards JB, et al. The effect of vilazodone on sexual function during the treatment of major depressive disorder. J Sex Med. 2013;10(10):2465–2476. PubMed
This PDF is free for all visitors!
![]() Save
Save
![]() Share
Share
![]() Cite
Cite