‘ ‹’ ‹
Click to enlarge page
Tardive dyskinesia (TD) is a disorder characterized by involuntary movements, typically of the orofacial muscles and also of the extremities and other muscle groups. The condition is associated with exposure to dopamine receptor blocking agents, including antipsychotics. Because the indications and off-label uses for these agents have expanded over the last 2 decades, a larger number of patients are receiving antipsychotic medications than in the past. While evidence suggests that patients being treated with second-generation antipsychotics have less risk for developing TD than those treated with first-generation antipsychotics, the decreased risk is not as great as was originally expected. In addition, patients with chronic psychiatric conditions often require long-term use of antipsychotics, putting them at risk for TD. This article addresses the prevalence, risk factors, and prevention of TD; assessment strategies including diagnostic criteria and rating scales; and evidence for TD treatments, including 2 newly approved medications: deutetrabenazine and valbenazine.
From the Department of Psyc’ ‹hiatry and Molecular Medicine, Hofstra Northwell School of Medicine, Hempstead; Center for Psychiatric Neuroscience, The Feinstein Institute for Medical Research, Manhasset; and Recognition and Prevention (RAP) Program, Department of Psychiatry, The Zucker Hillside Hospital, Glen Oaks, New York (Dr Correll); Department of Psychiatry, Hofstra Northwell School of Medicine, Hempstead; Center for Psychiatric Neuroscience, The Feinstein Institute for Medical Research, Manhasset; and Department of Psychiatry, The Zucker Hillside Hospital, Glen Oaks, New York (Dr Kane); and the Department of Psychiatry and Behavioral Sciences, New York Medical College, Valhalla (Dr Citrome).
‘ ‹’ ‹’ ‹

The teleconference was chaired by Christoph U. Correll, MD, Department of Psychiatry and Molecular Medicine, Hofstra Northwell School of Medicine, Hempstead; Center for Psychiatric Neuroscience, The Feinstein Institute for Medical Research, Manhasset; and Recognition and Prevention (RAP) Program, Department of Psychiatry, The Zucker Hillside Hospital, Glen Oaks, New York. The faculty were John M. Kane, MD, Department of Psychiatry, Hofstra Northwell School of Medicine, Hempstead; Center for Psychiatric Neuroscience, The Feinstein Institute for Medical Research, Manhasset; and Department of Psychiatry, The Zucker Hillside Hospital, Glen Oaks, New York; and Leslie L. Citrome, MD, MPH, Department of Psychiatry and Behavioral Sciences, New York Medical College, Valhalla.
Financial disclosure: Dr Correll is a consultant for and has received honoraria from Alkermes, Allergan, Gerson Lehrman Group, IntraCellular Therapies, Janssen/Johnson & Johnson, LB Pharma, Lundbeck, Medavante, Medscape, Neurocrine, Otsuka, Pfizer, Sunovion, Takeda, and Teva; has received grant/research support from Takeda; and has provided expert testimony for Bristol-Myers Squibb, Janssen, and Otsuka. Dr Kane has received grant/research support from Otsuka and Lundbeck; has received honoraria/consulting fees from Alkermes, Allergan, Forum, Intracellular Therapies, Johnson & Johnson, Janssen, LB Pharmaceuticals, Lundbeck, Minerva, Neurocrine, Otsuka, Pierre Fabre, Sunovion, Takeda, and Teva; and is a stock shareholder of MedAvante, Vanguard Research Group, and LB Pharmaceuticals. Dr Citrome is a consultant for Acadia, Alkermes, Allergan, Forum, IntraCellular Therapies, Janssen, Lundbeck, Merck, Neurocrine, Noven, Otsuka, Pfizer, Shire, Sunovion, Takeda, Teva, and Vanda; is a member of the speakers/advisory boards for Acadia, Alkermes, Allergan, Janssen, Lundbeck, Merck, Neurocrine, Otsuka, Pfizer, Shire, Sunovion, Takeda, and Vanda; and is a stock shareholder (small number of shares of common stock) of Bristol-Myers Squibb, Eli Lilly, Johnson & Johnson, Merck, and Pfizer.
The opinions expressed herein are those of the faculty and do not necessarily reflect the opinions of the CME provider and publisher or the commercial supporter.
J Clin Psychiatry 2017;78(8):1136-1147
https://doi.org/10.4088/JCP.tv17016ah4c
© Copyright 2017 Physicians Postgraduate Press, Inc.
Tardive dyskinesia (TD) is a condition characterized by involuntary movements of the face, torso, extremities, and sometimes the respiratory system. It is usually observed in patients after long-term treatment with antipsychotic agents.1 This extrapyramidal symptom was first described by Schonecker2 in 1957, a few years after the first antipsychotic agents were used.1 While evidence suggests that patients being treated with second-generation antipsychotics (SGAs) have less risk for developing TD than those treated with first-generation antipsychotics (FGAs), the decreased risk is not as great as was originally expected.3 Because FGAs are still in use and SGAs are being used for multiple illnesses, the potential development of TD among patients taking antipsychotics (or other agents that block dopamine receptors)4 needs to be monitored and, if it does occur, managed.
This Academic Highlights, based on a series of teleconferences given by Christoph U. Correll, MD (Chair); John M. Kane, MD; and Leslie L. Citrome, MD, MPH, will address the prevalence, risk factors, and prevention of TD; assessment strategies and tools; and evidence for TD treatments.
EPIDEMIOLOGY AND PREVENTION OF TARDIVE DYSKINESIA
Dr Correll discussed the clinical syndrome of TD, which consists of involuntary, repetitive movements that are mostly in the oral, lingual, and buccal regions. Common movements are tongue protrusion, puckering, chewing, and grimacing. Less commonly, abnormal involuntary movements occur in the hands, legs, feet, or torso. The condition can be severe and persistent and may have both medical and psychosocial consequences.3 Treatment with dopamine receptor blocking agents, such as antipsychotics, increases the risk for TD.3,5
In many cases, once TD is established, it cannot be reversed, and it has been associated with poorer quality of life6 and increased mortality.7 The indications and off-label uses for dopamine antagonist antipsychotic medications have expanded over the last 2 decades (including not only psychotic disorders but also mood disorders, such as bipolar disorder and unipolar depression, as well as anxiety disorders and insomnia).8 Therefore, a larger number of patients are receiving antipsychotic medications than in the past.
Dr Correll noted that the symptoms of TD are typically observed in patients who have received long-term treatment with antipsychotic medications, especially at higher doses associated with extrapyramidal symptoms (EPS), but TD may occur in untreated patients.9 Similar movement disorders had been described in patients with schizophrenia prior to the existence of dopamine antagonist medications, which could indicate that schizophrenia itself may be associated with aberrations in the dopamine system that may give rise to abnormal, involuntary movements in some patients.
Prevalence of Tardive Dyskinesia
Next, Dr Correll presented data on the prevalence of TD, while first considering some limitations in the literature.
Study limitations. When evaluating studies of the prevalence of TD, it is important to consider differences in study methodology. Retrospective chart reviews rely on clinical coding, which is often incomplete. Database studies include large samples of generalizable patients who are not required to provide informed consent, and patients can be followed for a long period of time. However, patient characteristics, treatment characteristics, and diagnostic evaluations of TD depend on clinical coding, which again is not always accurate. Cross-sectional prevalence studies provide a snapshot of a patient on a given treatment, but causality cannot be assigned. Prior treatments may result in TD even after they have been discontinued. Randomized controlled prospective studies compare different treatments and assess TD in a more or less detailed way. However, randomized trials rarely have TD as a primary focus, and only patients who agree to participate in a rigorous and often double-blind study are included. Prospective cohort studies are designed to assess TD and collect data about treatment effects and outcomes at predefined intervals, but this careful focus on TD might overemphasize its presence. Meta-analyses can pool available data but depend very much on the quality of the included studies.10

- Consider risk factors that could make patients more likely to develop TD.
- Seek to prevent TD by treating patients with the lowest dose of antipsychotics for the shortest duration necessary.
- Use TD diagnostic criteria combined with a rating scale (such as AIMS) assessment and differential diagnosis to determine if involuntary abnormal movements are caused by antipsychotic treatment.
- Consider treatments for TD based on evidence given in the American Academy of Neurology guidelines as well as the novel FDA-approved agents.
Ideally, generalizable samples would be used, which can be achieved by consecutive enrollment of patients. At different times and in different studies or regions, doses may be higher or lower, and dropout of patients in different treatment arms may vary greatly. These 2 factors—dosing differences and dropout rates—can affect observed rates of TD and need to be considered in the analysis and interpretation of the data. Similarly, whether TD is a primary or secondary outcome may affect sensitivity of the assessments, and the measurement and definition of TD will also affect outcomes. Furthermore, training, reliability of raters, and blinding differ across studies. Rating scale-based TD rates are more sensitive than spontaneous reports.
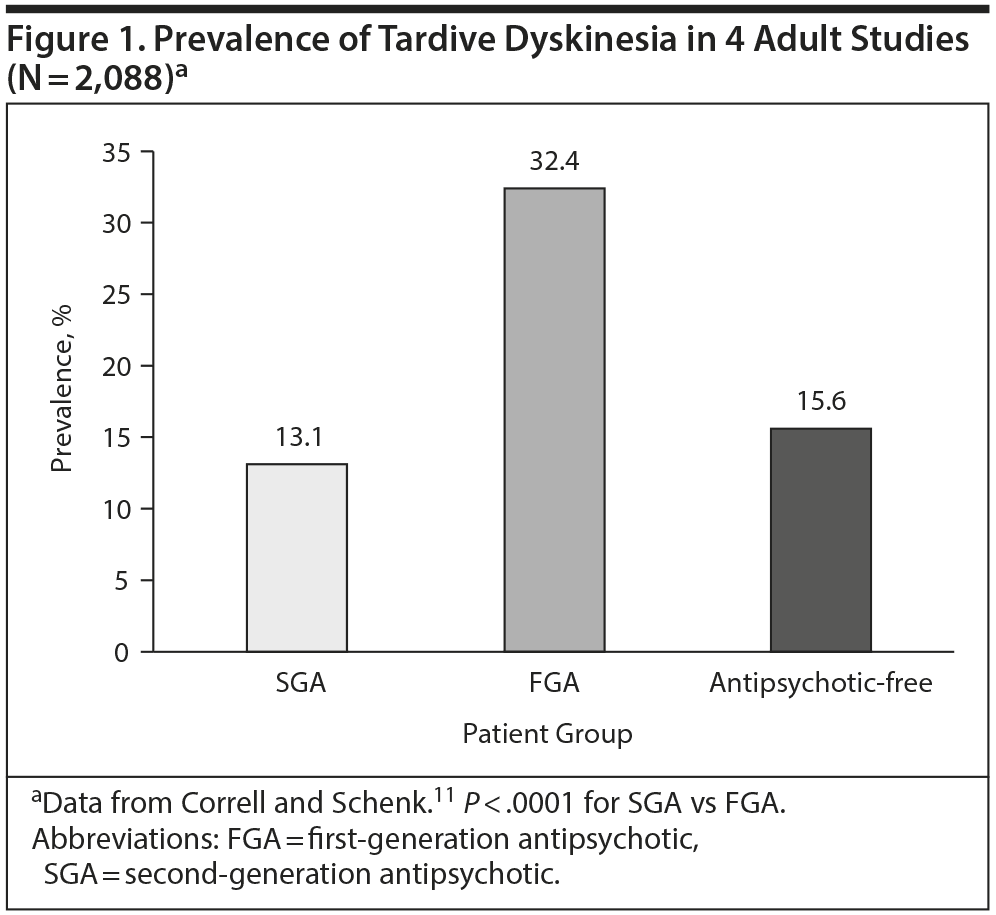
Prevalence data. A study by Correll and Schenk11 reviewed the prevalence of TD with FGAs and SGAs from 4 studies among adults between 2004 and 2008. Of the 2,088 patients, the mean age was 41.2 years, 71.2% were men, and 62.0% were white. The authors found that the prevalence of TD among patients taking SGAs was lower than for those taking FGAs and similar to that of patients who were not taking any antipsychotic at the time of assessment (Figure 1).11 Nevertheless, the TD prevalence rate with SGAs was higher than had previously been reported.
Click figure to enlarge
In a recent comprehensive meta-analysis, Carbon et al4 reviewed 41 studies from 2000 to 2015, a time period when both SGAs and FGAs were prescribed under similar conditions. These studies included 11,493 patients with a mean age of 42.8 years; 66.4% were male, and 77.1% had schizophrenia-spectrum disorders. The mean pooled TD rates were 20.7% with SGAs and 30.0% with FGAs (P = .002).
In the 20 studies that had at least 2 arms, one of which was an FGA arm and one of which was an SGA arm, the risk for TD was significantly lower with SGAs than with FGAs (P = .011).4
In the 9 studies in which an SGA and an FGA were combined, the risk of TD was lower with the combination than with an FGA alone (P < .001).4 This finding might suggest that combining an SGA with an FGA could potentially reduce TD rates seen with FGA use alone. Conversely, noted Dr Correll, it is also possible that the FGA was only added later and at a low dose to augment an SGA treatment that was ineffective. This strategy may have reduced the TD rate compared with higher-dose FGA monotherapy.
Four studies included patients who had never received an FGA; the TD prevalence rate with SGA-only exposure was 7.2%. The TD rate among SGA-treated patients who had probable previous FGA exposure (in 28 studies) was 23.4% (P < .001).4 This finding may suggest that prior FGA treatment could be responsible for at least a subgroup of patients who were found to have TD while being treated with SGAs. Nevertheless, due to the limitations mentioned above, cross-sectional studies need to be supplemented with carefully designed prospective studies to compare the risk directly between FGAs and SGAs.
A review12 from 2004 of studies of annual incidence rates of TD found a lower rate in adults taking various SGAs (0.8%) than in those taking an FGA (haloperidol; 5.4%). The same was true in pooled studies of non-elderly adults published between 2004 and 2008, with lower annual incidence rates of TD for SGAs than FGAs (2.98% vs 7.7%, P < .0001).11
Risk Factors for TD
Dr Correll also described risk factors for TD, which should help clinicians closely monitor specific patients for signs of TD. Risk factors include the following1:
- Older age
- Female sex
- African American ethnicity
- Preexisting mood disorder
- Cognitive disturbance
- Alcohol or substance abuse
- Higher dose/longer use of antipsychotic medications
- Treatment with typical neuroleptic (FGA) agents
- Use of lithium or antiparkinsonian agents
- Early occurrence of EPS
- Diabetes
- HIV positivity
Older age is consistently found to be a risk factor for TD.13 In the review of incidence studies that found a 0.8% annual incidence of TD in adults taking SGAs,12 a much higher incidence of TD was found in older patients. The 3 studies with older patients (mean age = 78.3 years) who were taking SGAs showed a pooled TD incidence rate of 5.3%.
In a separate study13 that investigated the incidence of TD in elderly patients being treated with FGAs, incidence rates were much higher (25% after 1 year of treatment, 34% after 2 years, and 53% after 3 years). These patients were 55 years or older when they started taking antipsychotics. Although most patients were being treated with low doses of FGAs, incidence rates of TD were 3 to 5 times higher than those found in studies of younger patients.13 A greater incidence of TD in this sample was associated with a history of electroconvulsive therapy, the presence of EPS early in treatment, and higher doses and greater cumulative antipsychotic exposure.13
Incidence of TD was also examined in a prospective study14 of older patients taking SGAs who had not previously taken FGAs. Patients were aged 55 years or older (mean age = 79.8 years). The cumulative TD rates after 1 year were 5.3% for risperidone and 6.7% for olanzapine, and the 2-year rates were 7.2% and 11.1%, respectively. The risk was greater in women, African Americans, and those with concurrent FGA treatment.
African American patients are at risk for developing TD. One study15 evaluated American outpatients with schizophrenia for TD over 1 to 10 years of follow-up. The European American group (n = 329) had significantly less TD than the African American group (n = 199; P < .05), despite controlling for differences in age and dosing.
While age and medication dose are well established risk factors, less established ones include cognitive impairment, mood disorders, preexisting movement disorders, alcohol and drug abuse, and diabetes.16
Case Practice Question
Case 1. Casey is a 31-year-old patient of European descent beginning antipsychotic treatment for bipolar disorder. He experiences parkinsonian symptoms during the up-titration to a dose that is close to the package insert maximum to control acute manic symptoms and agitation. Which of Casey’s characteristics is not an established risk factor for TD?
- Presence of acute EPS
- Younger age
- Mood disorder
- Higher antipsychotic dose
Discussion of Case Practice Question
Preferred response: b. Younger age
Older age is a very well established risk factor for TD while younger age is not. Because Casey is just 31, his age is not a risk factor. Bipolar disorder puts him at risk for TD since mood disorders are a risk factor. The dose of his antipsychotic medication is at the high end of indicated dosing, and his parkinsonian symptoms demonstrate early EPS, so these factors add to his risk for TD.
Prevention of TD
Because TD can be irreversible in some patients, Dr Correll provided some measures to limit the risk of TD (Table 1).17 Clinicians should follow the prescribing information for antipsychotic agents carefully. Long-term use of antipsychotic treatments should be considered only in cases in which discontinuing medication leads to worsening illness, such as chronic psychotic conditions. Dopamine antagonists should be prescribed at the lowest effective dose and for the shortest duration necessary.1,18 To avoid acute EPS, treatment should include dosing the medication carefully until efficacy is achieved and not increasing the dose more than is needed. The use of SGAs may lower the liability for EPS19 and TD compared with FGAs.4,12

Click figure to enlarge
Anticholinergic medications may worsen TD in some patients1 (although they may also be a marker of acute EPS that increases the TD risk). Clinicians should advise patients and their families about the risk for TD before starting an antipsychotic agent.1 Clinicians should also carefully monitor patients for abnormal movements using baseline and ongoing rating scale scores, such as the Abnormal Involuntary Movement Scale (AIMS),20 so that treatment can be changed if involuntary movements emerge.1
Conclusion
Dr Correll concluded that the prevalence of TD is still considerable despite predominant SGA use. Prevalence of TD is lower in patients currently taking SGAs versus FGAs and lower in patients taking SGAs without prior FGA treatment. Despite these findings, limitations in the literature hinder clinicians from getting an accurate picture of prevalence rates. The persistence, severity, and effect on quality of life of TD require further study. Clinicians should look for specific risk factors for TD in their patients, especially in the elderly, those taking high doses of antipsychotics, those using antipsychotics for longer durations, or those showing symptoms of acute EPS. Using preventive measures and careful monitoring, clinicians can help manage the risk of TD among patients who require treatment with antipsychotics and other dopamine receptor blocking agents.
ASSESSING PATIENTS FOR TARDIVE DYSKINESIA
Dr Kane outlined assessment strategies for TD. Because of the serious and potentially irreversible nature of TD, accurate diagnosis is crucial, but it can present a clinical challenge. The onset of TD is insidious, with fluctuating symptoms that can be masked by or confused with symptoms of other disorders or medication-induced side effects.21,22 Most commonly, TD will present as involuntary, repetitive, purposeless orofacial movements including chewing; lip smacking, puckering, or pursing; tongue protrusion; grimacing; or bulging of the cheeks.22-24 Patients may also experience contracting, twisting, or writhing movements of the fingers, hands, arms, or legs.22
Diagnostic Criteria
The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5),21 criteria for TD simply state that to receive this diagnosis an individual should exhibit involuntary movements lasting at least several weeks in association with neuroleptic medication use of at least a few months’ duration. The involuntary movements should be athetoid or choreiform and typically of the tongue, jaw, and extremities. Dr Kane and his colleague, Nina R. Schooler, PhD, developed a more in-depth set of diagnostic criteria for TD, known as the Schooler-Kane criteria,25 which were originally intended for research settings but have been widely used in clinical practice.22
The first of the 3 Schooler-Kane criteria is that the patient must have at least 3 months of antipsychotic exposure, which may be continuous or discontinuous. Second, the patient must exhibit abnormal, involuntary movements of moderate or greater severity in 1 or more body regions or mild severity in 2 or more body regions, according to a rating scale such as the AIMS. Third, the patient must be free of other conditions that may cause abnormal, involuntary movements. A patient who meets all 3 criteria has probable TD. Further evaluations are necessary to determine if movements improve, worsen, or stay the same; if dosing changed; and the length of time during which movements and medication have or have not been present.25 The rationales behind the Schooler-Kane criteria are presented below.
Antipsychotic Drug Exposure
Dr Kane explained that involuntary movements and EPS can be observed in untreated patients with schizophrenia. Drug-naive individuals with schizophrenia are significantly more likely to experience movement disorders such as dyskinesias compared with healthy controls (odds ratio = 3.59; 95% CI, 1.53-8.41).26 Dopamine dysregulation in specific brain regions may be involved in both the etiology of schizophrenia and the increased risk of movement disorders.26 Although the pathophysiology of TD is not fully understood, one hypothesis is that the antipsychotics’ dopamine receptor blocking action in combination with the altered striatal dopamine activity found in individuals with schizophrenia creates an imbalance between direct and indirect pathways of the basal ganglia, which are necessary for normal movement.24 Thus, clinicians should evaluate patients for the presence of involuntary movements before initiating antipsychotic treatment to ensure that any preexisting movements are not later incorrectly attributed to the medication.23 By requiring patients to have at least 3 months of cumulative antipsychotic exposure, the Schooler-Kane diagnostic criteria25 help clinicians confirm the association between medication and involuntary movement symptoms.
Rating Scales
To receive a diagnosis of TD according to the Schooler-Kane diagnostic criteria,25 a patient must exhibit either moderate involuntary movements in 1 or more body areas or at least mild involuntary movements in 2 or more body areas. Dr Kane specified that the presence of these movements should be determined by using a standardized rating scale,25 such as the Simpson Dyskinesia Scale,27 the Extrapyramidal Symptom Rating Scale (ESRS),28 or the AIMS.20
Clinicians must remember that these are screening and not diagnostic tools. Dr Kane noted that these instruments are useful for identifying signs and symptoms of involuntary movement and assessing the severity of these movements, but they should be used in conjunction with diagnostic criteria. Rating scales are also useful to facilitate communication between patients and clinicians regarding the adverse effects of treatment patients are experiencing. Patients may be reluctant or unable to share side effects voluntarily, and clinicians may underestimate the rate of side effects from antipsychotics in their patients.29 Consistent use of rating scales can help solve these problems by giving clinicians information on the severity, frequency, and impact of involuntary movement symptoms and by enabling patients to recognize and describe their symptoms.
The Simpson Dyskinesia Scale, specific for TD, was developed in both a long (34-item) and an abbreviated (13-item) version to help clinicians describe the breadth of TD syndrome and quantify the disorder.27
The ESRS28 is a 41-item scale plus 4 global impression questions to assess the types of drug-induced movement disorders: parkinsonism, akathisia, dystonia, and TD. In a cross-scale comparison, the ESRS and AIMS were found to have a 96% (359/374) agreement between TD cases defined by DSM criteria.28
The AIMS was developed by the National Institute of Mental Health as a research tool, but because of its utility for identifying the abnormal, involuntary movements characteristic of TD, it has become widely recommended for use in clinical practice to screen for the involuntary movements required to diagnose TD using the Schooler-Kane criteria.23 The 12-item AIMS assesses 10 items on a 5-point scale (0-4: none, minimal, mild, moderate, severe) in different parts of the body, including the face, extremities, and trunk, as well as global assessment items addressing overall severity, impact, and awareness of abnormal movements. The final 2 yes or no questions address problems with teeth or dentures. Items 1 through 7 quantify the different abnormal movements that can be evident in the face, extremities, and trunk. These 7 items are usually summed to form a total dyskinesia score.
Although the AIMS is a useful tool, Dr Kane pointed out that clinicians must administer the scale properly in order to obtain accurate and consistent results.30 The clinician cannot simply observe the patient for a few seconds or a minute. Ideally, the clinician should attempt to discreetly observe the patient at rest, perhaps while he or she is in the waiting room, and also should look for involuntary movements when first greeting the patient.
Once in the office, a more systematic examination can take place, and a firm chair will be needed. Dr Kane recommended that the chair have either no arms or arms that are short enough for the patient’s hands to hang over the ends. The clinician must ask the patient if anything is in his or her mouth (like chewing gum) and if so to remove it, about any problems with his or her teeth, and whether dentures are being worn.23 The clinician should also inquire about the patient’s awareness of any involuntary movement.
As the AIMS examination proceeds, the clinician will continually look for any abnormal movements as the patient performs a variety of different movements. The patient should sit in the chair with hands on knees, legs slightly apart, and feet flat on the floor. Then the patient should sit with his or her arms and hands unsupported.
The clinician should ask the patient to open his or her mouth so that the clinician can observe the tongue at rest, and then the patient should be asked to protrude the tongue and keep it extended. Repeating these tongue observations later in the evaluation may be beneficial, noted Dr Kane.
The patient should be asked to rapidly tap his or her thumb to each finger on that hand for 10 to 15 seconds, and then switch to the other hand, while the clinician observes the entire body for any abnormal involuntary movements that may be elicited. The clinician should ask the patient to rise from the chair, walk, and then extend his or her arms in front with palms down, while continually observing the patient’s entire body.
When scoring the AIMS, the clinician should record the incidence of a movement regardless of whether the movement was activated by the requested actions. That is, some clinicians have assigned a lower score to movements that did not appear until they asked the patient to perform an action, and they assigned a higher score to movements that existed when the patient was at rest. Instead, Dr Kane recommended that all movements should be rated in the same way. An AIMS score of at least 2 in 2 or more body regions or a score of 3 or 4 in ≥ 1 body region is used for a probable diagnosis of TD with the Schooler-Kane criteria.25
According to the American Psychiatric Association (APA),31 all patients taking an antipsychotic medication should be regularly monitored for movement disorders. The APA recommends that patients taking FGAs be assessed every 6 months, whereas patients taking SGAs should be assessed every 12 months. If patients have risk factors for TD (eg, older age, presence of an affective disorder1), they should be monitored more frequently. Patients at increased risk who are receiving an FGA should be assessed every 3 months, and patients at increased risk who are receiving an SGA should be assessed every 6 months.31 If a patient has recently initiated antipsychotic treatment and early involuntary movement has been detected, this patient should be considered at risk for TD and monitored more frequently to determine if the symptoms persist long enough (ie, 3 months) to meet the criteria for TD.1,31
Case Practice Question
Case 2. Beverly is a 68-year-old new patient with treatment-resistant major depressive disorder (MDD). Two months ago, she had an SGA added to her third antidepressant treatment. Beverly was referred because her MDD has improved but not quite remitted. Her referring physician had administered the AIMS before adding the SGA, and she had no involuntary movements. You administer the AIMS and find mild involuntary lip movements. Which of the following next steps would be most appropriate?
- Diagnose Beverly with probable TD
- Consider Beverly to be at risk for TD and assess her again in 6 months
- Consider Beverly to be at risk for TD and assess her again in 3 months
- Consider Beverly to be at risk for TD and assess her again in 1 month
Discussion of Case Practice Question
Preferred response: c. Consider Beverly to be at risk for TD and assess her again in 3 months
Beverly has an AIMS score of 2 (mild) in one body region. According to Schooler-Kane criteria, Beverly should not be diagnosed with probable TD because her cumulative antipsychotic use is less than 3 months and her involuntary movements are not severe enough or widespread enough. But, because she has a higher risk for TD (being elderly and female and having a mood disorder) and has developed early involuntary movements, the best option for the clinician is to reassess her when she has had 3 months of antipsychotic exposure.
Dr Kane emphasized that before diagnosing TD, the clinician must rule out other possible conditions that might be producing the involuntary movements.25 Patients who develop dyskinesias after starting antipsychotic treatment should be evaluated for clues to a cause for the dyskinesia, such as family history, sudden onset versus progressive course, or associated neurologic or medical abnormalities.22 Once neurologic causes have been eliminated, numerous differential diagnoses must be considered. Conditions that may resemble TD include spontaneous dyskinesias (ie, not associated with antipsychotic exposure), various syndromes, Huntington disease, and Wilson disease (Table 2).22,23

Click figure to enlarge
Additional conditions that may resemble TD are other movement disorders known collectively by the term tardive syndromes, which include tardive dystonia (constant or recurrent muscle contractions), tardive akathisia (restless or jittery feeling), tardive stereotypy (repetitive coordinated movements that may look purposeful), tardive myoclonus (quick, uncontrollable muscle jerks), tardive tourettism or tics (repeated brief movements or sounds), or tardive tremor (shaking movements).16,24 While some of these syndromes may occur simultaneously with TD, they have distinct clinical characteristics from TD. For example, tardive stereotypy may present as abnormal movement in the oro-buccal-facial region similar to TD, but the movements are more predictable, continuous, and are typically distractible. Tardive dystonia is more common in young male patients, while TD is more common in the elderly and women. Tardive akathisia is characterized by an inner restlessness, causing an inability to sit or stand still. Patients with tardive akathisia may shift from foot to foot, walk in place, rock their bodies, or cross/uncross their legs.24
Dr Kane noted that clinicians must also rule out whether the movements are caused by drugs or substances known to be associated with dyskinesias, such as caffeine, antihistamines, stimulants, phenytoin, estrogens, or antidepressants, or even simply caused by oral problems or ill-fitting dentures.22,23 Once all of these possible explanations for involuntary movements have been eliminated, the patient meets criteria for a diagnosis of probable TD.25
Conclusion
Although the sometimes subtle and fluctuating symptoms of TD can be difficult to identify, Dr Kane stated that clinicians can improve their recognition of this disorder through careful monitoring and consistent use of diagnostic criteria and rating scales. The Schooler-Kane criteria combined with a properly administered rating scale, such as the AIMS, will enable clinicians to increase early recognition of TD.
COMPARING TREATMENTS FOR TARDIVE DYSKINESIA
Once TD has been recognized in a patient, what are the treatment options? Dr Citrome examined the evidence for various treatments for TD. Different treatments have been assessed for the management of TD, with studies typically using the AIMS to measure symptoms.20
Treatment Circa 2007
For a long time, the treatment of TD was an unmet clinical need, with few options having strong evidence of efficacy without substantial side effect risks. Dr Citrome explained that, a decade ago, one treatment strategy for TD would be to gradually switch a patient from an FGA to an SGA (other than clozapine) and discontinue any anticholinergic medications.23,32 If TD improved, the SGA was continued. The next step, if TD persisted, was to switch to a second SGA other than clozapine. If TD remained, the next step was to switch to clozapine. If TD continued after the above steps were taken, suppression therapy was often considered and usually consisted of the combination of an FGA and an SGA.
Other treatment options included tetrabenazine, which is discussed later, or branched-chain amino acids (BCAAs). The BCAAs were studied as a possible intervention for TD because of the observed association between dyskinetic movements and impaired clearance of phenylalanine. It is thought that ingesting BCAAs decreases the availability of phenylalanine to the brain; thus, BCAAs might improve TD by decreasing amine neurotransmitter synthesis.33,34
A 3-week randomized double-blinded study34 compared high-dose BCAAs with placebo in men with TD (n = 36). The dyskinetic movements decreased by a mean of 36.5% in the BCAA group but increased by a mean of 3.4% in the placebo group. One-third of the BCAA group had a reduction of 60% or more in dyskinetic movements, compared with none of the subjects receiving placebo.34
A combination of 3 BCAAs was approved by the US Food and Drug Administration (FDA) as a medical food for the dietary management of TD in male patients.35 The product was a flavored powder to mix with water and then drink 3 times daily.33 However, the drink mix was not calorie free and was unpalatable for some. Although the branded product is no longer available, compounding pharmacies can make it using the same ratio of ingredients (L-leucine, L-valine, and L-isoleucine) tested in the clinical trial.34
Proposed treatments for TD also included donepezil, melatonin, vitamin B6, and vitamin E, which had mixed results in clinical trials.23,32
2013 Recommendations From the American Academy of Neurology
Moving ahead several years, Dr Citrome discussed the 2013 American Academy of Neurology (AAN) evidence-based guidelines for the treatment of TD.36 The AAN determined that data were insufficient as to whether stopping the medication associated with TD would suppress symptoms.36,37 The strategy of switching from an FGA to an SGA was also determined to have insufficient evidence. Raising the antipsychotic dose may slightly reduce symptom severity, but the symptoms may be masked, and a temporary improvement could lead to ever-increasing doses.38,39 Insufficient data to support (or refute) their use was found for acetazolamide, bromocriptine, thiamine, baclofen, vitamin E, vitamin B6, selegiline, melatonin, nifedipine, levetiracetam, buspirone, yi-gan san, biperiden discontinuation, botulinum toxin type A, electroconvulsive therapy, α-methyldopa, reserpine, and pallidal deep brain stimulation. The guidelines did not recommend diltiazem, galantamine, and eicosapentaenoic acid.
According to the guidelines,36 the following interventions have some evidence to support their use: clonazepam, Ginkgo biloba, amantadine, and tetrabenazine.
Clonazepam. The antianxiety agent, clonazepam, is a benzodiazepine with indirect GABA agonist activity and has been explored as a treatment for TD since the 1970s.3 A 1981 study40 compared clonazepam with phenobarbital in 21 psychiatric patients with TD. While both agents reduced dyskinetic movements, clonazepam showed greater efficacy for orofacial dyskinesia while phenobarbital was more effective for abnormal limb and axial movements.
A decade later, clonazepam was assessed in a 12-week, double-blind, placebo-controlled, randomized crossover study41 with an open-label, 9-month extension. Of the 19 chronically ill patients with TD, 11 received 4.0 to 4.5 mg/d of clonazepam, 6 received 3.0 mg/d, and 2 received 2.0 mg/d. Treatment alternated between active medication and placebo. The primary efficacy endpoint was change in dyskinesia score using the Maryland Psychiatric Research Center Movement Disorder Scale as assessed by blinded video raters. Clonazepam treatment reduced dyskinesia scores by approximately 37%, an effect that was reversed during placebo administration. However, tolerance developed in the 5 patients who received clonazepam in the long-term extension after 5-8 months of use.41
Ginkgo biloba. Extract of Ginkgo biloba (EGb), a potent antioxidant with free-radical scavenging activity, was assessed in a meta-analysis42 of 3 12-week, randomized-controlled trials in schizophrenia patients with TD (n = 299) in China. The primary outcome measure was severity of TD symptoms measured by the AIMS. Results showed that adjunctive EGb (240 mg/d) reduced TD symptoms compared with patients taking antipsychotic monotherapy (P < .00001). Discontinuation rates were similar between the EGb group and control group.
Amantadine. Amantadine blocks N-methyl-d-aspartate receptors.3 Amantadine was assessed in 2 double-blind, placebo-controlled, crossover studies.43,44 In a study by Pappa and colleagues,43 22 patients were treated with amantadine (up to 400 mg/d) or placebo, along with antipsychotic medications. With amantadine, the average total AIMS score reduction was 22%, compared with no reduction with placebo.43 In the other study,44 16 patients were treated with amantadine (300 mg/d) or placebo, as well as their antipsychotic medication. With amantadine, 4 patients’ AIMS scores increased, but overall, a mean 15% reduction was found, which was significant compared with placebo (P < .05).44
Tetrabenazine. Tetrabenazine was originally developed in the 1950s and approved in the United States in 2008 as an orphan drug for the treatment of choreiform movements associated with Huntington disease.39,45 Tetrabenazine is a reversible and specific inhibitor of vesicular monoamine transporter 2 (VMAT2), a protein in the brain that transports monoamine neurotransmitters (ie, dopamine, norepinephrine, serotonin, histamine) into vesicles for release into the synapse.46 The reduction of synaptic dopamine levels limits overstimulation and reduces the symptoms of involuntary movements associated with Huntington disease, Tourette syndrome, and TD.
The first TD study with tetrabenazine47 was published in 1972. After a 4-week placebo period, 24 inpatients were placed on tetrabenazine for 6 weeks. The antidyskinetic effect was marked in two-thirds of the patients. Another study48 compared haloperidol use with tetrabenazine use in 13 subjects and found that tetrabenazine produced significant reduction in the frequency of oral dyskinesia, with almost complete suppression in 2 patients. In a single-blind study49 that compared tetrabenazine efficacy using a randomized videotape protocol pre- and posttreatment, a 54% mean improvement on the AIMS motor subset scores was found.49 The above short-term results for tetrabenazine are further supported by long-term observational studies.36
Despite these positive results, tetrabenazine has limiting adverse effects—somnolence, parkinsonism, insomnia, and akathisia—as well as a short half-life (requiring 2-3 doses/d) and potential drug-drug interactions.39,50,51 These effects are related to the pharmacokinetics and pharmacodynamics of tetrabenazine, which is a one-to-one mixture of enantiomers.52 The α and β enantiomers each give rise to 2 isomers of a dihydrotetrabenazine metabolite for a total of 4 isomers. Those derived from the α-tetrabenazine isomer are active VMAT2 inhibitors and contribute to the therapeutic effects of the drug. These are metabolized via cytochrome P450 2D6 and 3A4 isoenzymes. The 2 derivatives of β-tetrabenazine are antagonists at the dopamine D2 receptor and can induce sedation and parkinsonism.52 These β-tetrabenazine derivatives are metabolized solely by CYP2D6; thus, side effects are more pronounced in the presence of CYP2D6 inhibitors. Consequently, for tetrabenazine doses above 50 mg/d, genotyping for the cytochrome P450 2D6 enzyme is required.51
The product label for tetrabenazine51 also carries a warning for depression (possibly related to serotonin depletion46) and suicide risk.
New VMAT2 Inhibitors
Due to the limitations of prior TD treatments, other agents have been explored as alternatives. Since the AAN guidelines were published, the US Food and Drug Administration (FDA) acknowledged the need for new treatment options with breakthrough therapies.52,53 Novel agents include valbenazine and deutetrabenazine, which are both reversible VMAT2 inhibitors.54 Valbenazine was approved as treatment for TD by the FDA in April 2017.55 Deutetrabenazine was FDA-approved for the treatment of TD in August 2017.56
Valbenazine. Valbenazine is metabolized slowly to a potent, selective VMAT2 antagonist, which enables once-daily dosing, does not require genotyping, and provides efficacy for TD as shown in placebo-controlled studies.57
A 6-week, double-blind, placebo-controlled phase 2 trial58 examined valbenazine (25-75 mg/d) in 102 patients with moderate-to-severe TD. These patients’ TD was associated with the use of neuroleptics or metoclopramide, taken for schizophrenia, schizoaffective disorder or mood disorders, or gastrointestinal disorders, respectively. The primary outcome was change on the AIMS (total score on items 1-7) from baseline to week 6 as assessed by blinded video raters who were movement disorder specialists otherwise not involved in the study. The once-daily starting dose of valbenazine was 25 mg, which could be escalated by 25 mg every 2 weeks up to 75 mg/d. After titration, 76% of those taking valbenazine reached the 75-mg/d dose (9 reached 50 mg/d, and 5 maintained 25 mg/d). The least-squares mean change in AIMS dyskinesia scores from baseline to endpoint was significantly greater with valbenazine (-2.6) than placebo (-0.2; P = .0005). No patients discontinued because of an adverse event.58
Click figure to enlarge
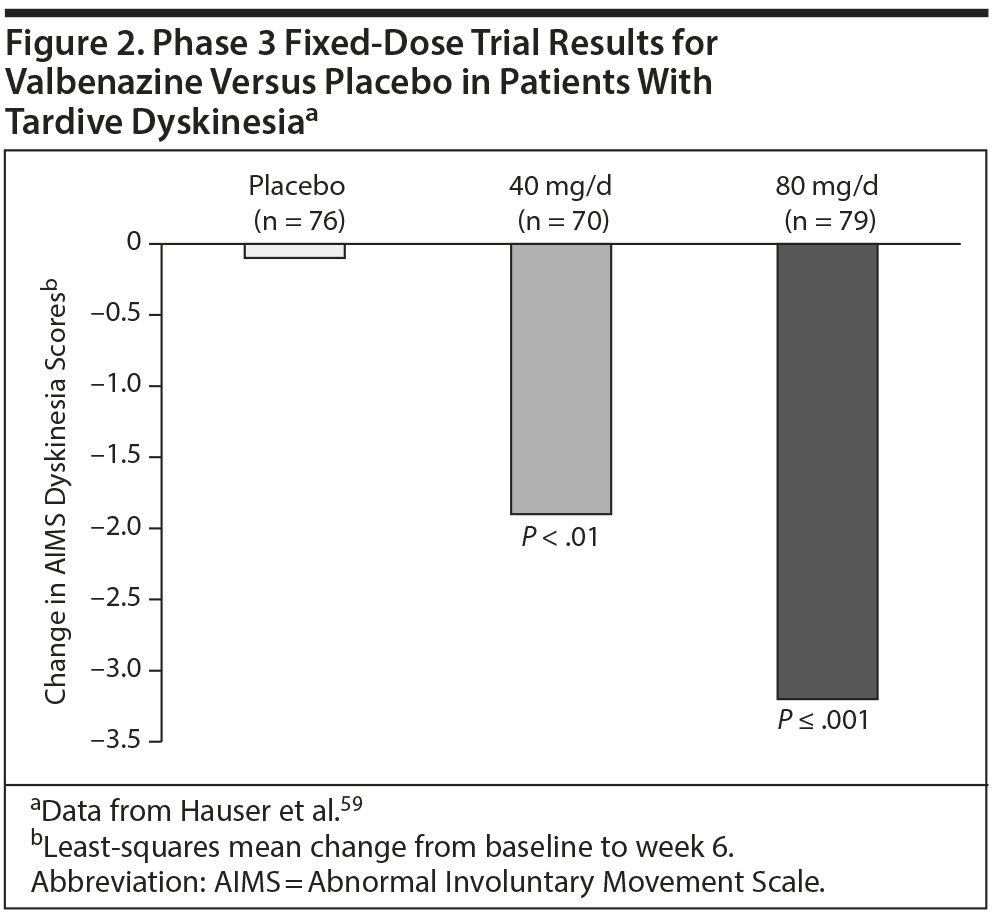
A 6-week, double-blind, randomized phase 3 trial59 compared fixed-dose valbenazine (40 and 80 mg/d) with placebo in patients with schizophrenia, schizoaffective disorder, or a mood disorder and TD (N = 234). The primary efficacy endpoint was change in AIMS dyskinesia scores (assessed by blinded video raters) from baseline to week 6 in the 80 mg/d group versus the placebo group. Valbenazine reduced AIMS dyskinesia scores, with the least-squares mean change from baseline to endpoint significantly greater for 80 mg/d versus placebo (P ≤ .001; Figure 2).59 The AIMS dyskinesia score was also reduced in the 40 mg/d group compared with placebo (-1.9 vs -0.1; P < .01). In terms of robust response (ie, ≥ 50% reduction in AIMS dyskinesia score from baseline), the percentage of participants responding was significantly higher in the 80 mg/d group than placebo at all study visits. At week 6, 40% of the 80-mg group had responded, compared with 8.7% in the placebo group.59 Discontinuation because of an adverse event occurred in 3.8% of those taking valbenazine (both doses pooled) versus 2.6% of those taking placebo.60
Product labeling for valbenazine recommends starting at 40 mg once daily with or without food and, after 1 week, increasing the dose to 80 mg once daily. The most common adverse reaction (at ≥ 5% and twice the rate of placebo) is somnolence (11% vs 4%).55 Additionally, there was no difference in low incidences of depression or suicidality between valbenazine and placebo.
Case Practice Question
Case 3. Andy, your 52-year-old patient, has received antipsychotics on and off for the past 30 years for the treatment of schizophrenia. When examined, he appears to be grimacing, and when he opens his mouth, his tongue sometimes sticks out. You administer the AIMS, and his total score on items 1-7 is 12. In terms of best evidence, which of the following treatments could be offered?
- Vitamin B6 and/or vitamin E
- Galantamine
- Ginkgo biloba
- An FDA-approved VMAT2 inhibitor
Discussion of Case Practice Question
Preferred response: Preferred response: d. An FDA-approved VMAT2 inhibitor
According to American Academy of Neurology Guidelines,36 Ginkgo biloba has some evidence supporting its use. Vitamins E and B6 do not have enough evidence to support or refute their efficacy in TD, while galantamine was not recommended. However, since the guidelines were published in 2013, novel VMAT2 inhibitors have been approved by the FDA for TD based on sufficient evidence of their efficacy.
Deutetrabenazine. Deutetrabenazine is related to tetrabenazine in that deuterium is substituted for hydrogen at the sites of primary metabolism, with improvements in tolerability aided by slower drug metabolism.61 Deuterium is a stable, nonradioactive, nontoxic, naturally occurring isotope of hydrogen and has the same size and shape as a hydrogen atom, differing only in forming stronger chemical bonds. Deutetrabenazine does not require genotyping. Efficacy of deutetrabenazine for TD was shown in placebo-controlled studies as outlined below.
In a 12-week, double-blind, phase 2 trial,61 117 patients with TD were randomly assigned to take twice-daily placebo or twice-daily deutetrabenazine. Dose was started at 6 mg twice daily and titrated weekly by 6 mg/d, if required, for up to 6 weeks until TD was controlled, a significant adverse event occurred, or the maximum dose (48 mg/d) was achieved. At the end of the titration period, the mean total daily dose was 38.8 mg/d. By endpoint, least-squares mean change from baseline in AIMS scores was significantly greater for the deutetrabenazine group versus the placebo group (-3.0 vs -1.6, respectively; P = .019). Fewer patients discontinued because of an adverse event with deutetrabenazine than with placebo (1.7% vs 3.4%, respectively). In addition, the incidences of depression/depressed mood and suicidal ideation in the deutetrabenazine group were similar to or lower than those in the placebo group.61
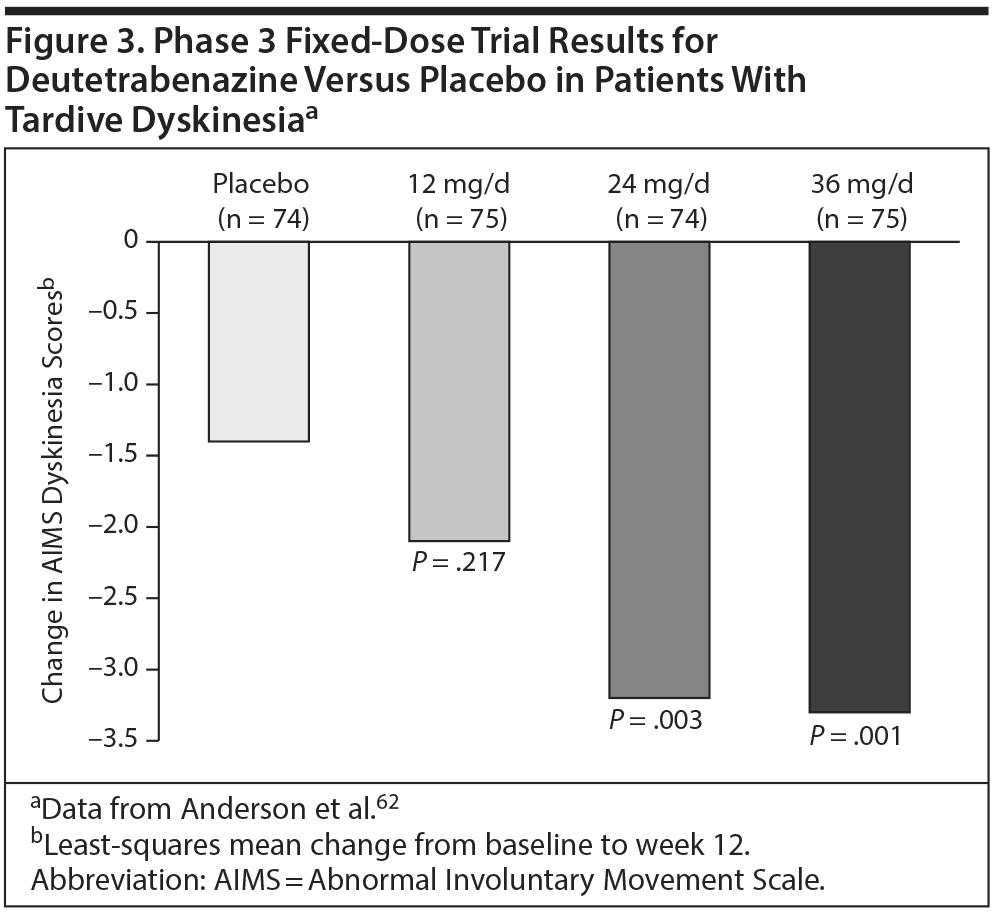
In a 12-week phase 3 trial,62 298 patients with TD were randomly assigned to receive deutetrabenazine (12 mg, 24 mg, or 36 mg daily in 2 divided doses) or placebo. Patients taking deutetrabenazine started at 12 mg daily and then gradually increased over 4 weeks until the randomized dose was reached; the doses were maintained over 8 weeks. The AIMS improvement from baseline to endpoint was significantly (P < .05) greater for those taking 24 or 36 mg/d of deutetrabenazine than for those taking placebo (Figure 3).62 The percentages of patients discontinuing because of an adverse event were 4% for deutetrabenazine (all doses pooled) versus 3% for placebo.62 Low incidences of depression and suicidal ideation were observed.62
Click figure to enlarge
Per product labeling,56 deutetrabenazine dose is determined individually for each patient based on reduction of TD and tolerability. The recommended starting dose of deutetrabenazine for TD is 6 mg twice daily, administered with food, and the dose can be increased at weekly intervals in increments of 6 mg/d to a maximum recommended daily dosage of 48 mg. The most common adverse reactions in patients with TD (at ≥ 4% and greater than the rate of placebo) were nasopharyngitis and insomnia (4% vs 2%, and 4% vs 1%, respectively).
Dr Citrome noted that, while the efficacy and tolerability of these new VMAT2 inhibitors are encouraging, one consideration that clinicians have with the use of new agents is their cost.60 Depending on insurance and health care setting, costs will vary for patients.
Conclusion
Dr Citrome concluded that patients taking antipsychotics must be monitored as recommended using the AIMS and should be treated promptly if TD is present. Although the VMAT2 inhibitor tetrabenazine shows promise for alleviating TD, it has several limitations. Two novel VMAT2 inhibitors have been developed to retain the efficacy of tetrabenazine but offer improved safety and tolerability. These agents, valbenazine and deutetrabenazine, have been FDA-approved for the treatment of TD and could provide much-needed treatment options to alleviate TD in patients who require continued treatment with antipsychotics for their chronic conditions.
Published online: October 10, 2017.
Disclosure of off-label usage: The authors have determined that, to the best of their knowledge, valbenazine and deutetrabenazine are the only drugs approved by the US Food and Drug Administration for the treatment of tardive dyskinesia.
Find more articles on this and other psychiatry and CNS topics:
The Journal of Clinical Psychiatry
The Primary Care Companion for CNS Disorders
REFERENCES
1. Jankelowitz SK. Treatment of neurolept-induced tardive dyskinesia. Neuropsychiatr Dis Treat. 2013;9:1371-1380. PubMed CrossRef
2. Schonecker M. Paroxysmal dyskinesia as the effect of megaphen. (German) Nervenarzt. 1957;28(12):550-553. PubMed
3. Lerner PP, Miodownik C, Lerner V. Tardive dyskinesia (syndrome): current concept and modern approaches to its management. Psychiatry Clin Neurosci. 2015;69(6):321-334. PubMed CrossRef
4. Carbon M, Hsieh C-H, Kane JM, et al. Tardive dyskinesia prevalence in the period of second-generation antipsychotic use: a meta-analysis. J Clin Psychiatry. 2017;78(3):e264-e278. PubMed CrossRef
5. Mehta SH, Morgan JC, Sethi KD. Drug-induced movement disorders. Neurol Clin. 2015;33(1):153-174. PubMed CrossRef
6. Browne S, Roe M, Lane A, et al. Quality of life in schizophrenia: relationship to sociodemographic factors, symptomatology and tardive dyskinesia. Acta Psychiatr Scand. 1996;94(2):118-124. PubMed CrossRef
7. Ballesteros J, González-Pinto A, Bulbena A. Tardive dyskinesia associated with higher mortality in psychiatric patients: results of a meta-analysis of seven independent studies. J Clin Psychopharmacol. 2000;20(2):188-194. PubMed CrossRef
8. Carton L, Cottencin O, Lapeyre-Mestre M, et al. Off-label prescribing of antipsychotics in adults, children and elderly individuals: a systematic review of recent prescription trends. Curr Pharm Des. 2015;21(23):3280-3297. PubMed CrossRef
9. Lerner V, Miodownik C. Motor symptoms of schizophrenia: is tardive dyskinesia a symptom or side effect? a modern treatment. Curr Psychiatry Rep. 2011;13(4):295-304. PubMed CrossRef
10. Correll CU, Rubio JM, Inczedy-Farkas G, et al. Efficacy of 42 pharmacologic cotreatment strategies added to antipsychotic monotherapy in schizophrenia: systematic overview and quality appraisal of the meta-analytic evidence. JAMA Psychiatry. 2017;74(7):675-684. PubMed CrossRef
11. Correll CU, Schenk EM. Tardive dyskinesia and new antipsychotics. Curr Opin Psychiatry. 2008;21(2):151-156. PubMed CrossRef
12. Correll CU, Leucht S, Kane JM. Lower risk for tardive dyskinesia associated with second-generation antipsychotics: a systematic review of 1-year studies. Am J Psychiatry. 2004;161(3):414-425. PubMed CrossRef
13. Woerner MG, Alvir JM, Saltz BL, et al. Prospective study of tardive dyskinesia in the elderly: rates and risk factors. Am J Psychiatry. 1998;155(11):1521-1528. PubMed CrossRef
14. Woerner MG, Correll CU, Alvir JMJ, et al. Incidence of tardive dyskinesia with risperidone or olanzapine in the elderly: results from a 2-year, prospective study in antipsychotic-naive patients. Neuropsychopharmacology. 2011;36(8):1738-1746. PubMed CrossRef
15. Wonodi I, Adami HM, Cassady SL, et al. Ethnicity and the course of tardive dyskinesia in outpatients presenting to the motor disorders clinic at the Maryland psychiatric research center. J Clin Psychopharmacol. 2004;24(6):592-598. PubMed CrossRef
16. Waln O, Jankovic J. An update on tardive dyskinesia: from phenomenology to treatment. Tremor Other Hyperkinet Mov (N Y). 2013;3:tre-03-161-4138-1. PubMed
17. Tarsy D, Lungu C, Baldessarini RJ. Epidemiology of tardive dyskinesia before and during the era of modern antipsychotic drugs. Handb Clin Neurol. 2011;100:601-616. PubMed CrossRef
18. Zádori D, Veres G, Szalárdy L, et al. Drug-induced movement disorders. Expert Opin Drug Saf. 2015;14(6):877-890. PubMed CrossRef
19. Divac N, Prostran M, Jakovcevski I, et al. Second-generation antipsychotics and extrapyramidal adverse effects. BioMed Res Int. 2014;2014:656370. PubMed CrossRef
20. Guy W. ECDEU Assessment Manual for Psychopharmacology. Washington, DC: US Department of Health, Education, and Welfare; 1976.
21. American Psychiatric Association. Diagnostic and Statistical Manual for Mental Disorders. Fifth Edition. Washington, DC: American Psychiatric Association; 2013.
22. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148. PubMed CrossRef
23. Citrome L, Dufresne R, Dyrud JM. Tardive dyskinesia: minimizing risk and improving outcomes in schizophrenia and other disorders. Am J Manag Care. 2007. http://www.ajmc.com/journals/supplement/2007/2007-12-vol12-n1-decisionmakernews/dec07-2760p1-12. Accessed December 20, 2016.
24. Aquino CCH, Lang AE. Tardive dyskinesia syndromes: current concepts. Parkinsonism Relat Disord. 2014;20(suppl 1):S113-S117. PubMed CrossRef
25. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486-487. PubMed CrossRef
26. Koning JPF, Tenback DE, van Os J, et al. Dyskinesia and parkinsonism in antipsychotic-naive patients with schizophrenia, first-degree relatives and healthy controls: a meta-analysis. Schizophr Bull. 2010;36(4):723-731. PubMed CrossRef
27. Simpson GM, Lee JH, Zoubok B, et al. A rating scale for tardive dyskinesia. Psychopharmacology (Berl). 1979;64(2):171-179. PubMed CrossRef
28. Chouinard G, Margolese HC. Manual for the Extrapyramidal Symptom Rating Scale (ESRS). Schizophr Res. 2005;76(2-3):247-265. PubMed CrossRef
29. Stomski NJ, Morrison P, Meyer A. Antipsychotic medication side effect assessment tools: a systematic review. Aust N Z J Psychiatry. 2016;50(5):399-409. PubMed CrossRef
30. Bark N, Florida D, Gera N, et al. Evaluation of the routine clinical use of the Brief Psychiatric Rating Scale (BPRS) and the Abnormal Involuntary Movement Scale (AIMS). J Psychiatr Pract. 2011;17(4):300-303. PubMed CrossRef
31. Lehman AF, Lieberman JA, Dixon LB, et al. Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry. 2004;161(2 suppl):1-56. PubMed
32. Margolese HC, Chouinard G, Kolivakis TT, et al. Tardive dyskinesia in the era of typical and atypical antipsychotics, part 2: incidence and management strategies in patients with schizophrenia. Can J Psychiatry. 2005;50(11):703-714. PubMed CrossRef
33. Roth LS. Tarvil for tardive dyskinesia: are we robbing Peter to pay Paul? Fed Pract. 2004;21(11):48.
34. Richardson MA, Bevans ML, Read LL, et al. Efficacy of the branched-chain amino acids in the treatment of tardive dyskinesia in men. Am J Psychiatry. 2003;160(6):1117-1124. PubMed CrossRef
35. North America SHS. First medical food treatment for antipsychotic drug-related tardive dyskinesia introduced. PR Newswire. http://www.prnewswire.com/news-releases/first-medical-food-treatment-for-anti-psychotic-drug-related-tardive-dyskinesia-introduced-77571632.html. Published May 22, 2002. Accessed June 15, 2017.
36. Bhidayasiri R, Fahn S, Weiner WJ, et al. Evidence-based guideline: treatment of tardive syndromes: report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2013;81(5):463-469. PubMed CrossRef
37. Zutshi D, Cloud LJ, Factor SA. Tardive syndromes are rarely reversible after discontinuing dopamine receptor blocking agents: experience from a university-based movement disorder clinic. Tremor Other Hyperkinet Mov (N Y). 2014;4:266. PubMed CrossRef
38. Mentzel CL, Bakker PR, van Os J, et al. Effect of antipsychotic type and dose changes on tardive dyskinesia and parkinsonism severity in patients with a serious mental illness: the Curaçao Extrapyramidal Syndromes Study XII. J Clin Psychiatry. 2017;78(3):e279-e285. PubMed CrossRef
39. Cloud LJ, Zutshi D, Factor SA. Tardive dyskinesia: therapeutic options for an increasingly common disorder. Neurotherapeutics. 2014;11(1):166-176. PubMed CrossRef
40. Bobruff A, Gardos G, Tarsy D, et al. Clonazepam and phenobarbital in tardive dyskinesia. Am J Psychiatry. 1981;138(2):189-193. PubMed CrossRef
41. Thaker GK, Nguyen JA, Strauss ME, et al. Clonazepam treatment of tardive dyskinesia: a practical GABAmimetic strategy. Am J Psychiatry. 1990;147(4):445-451. PubMed CrossRef
42. Zheng W, Xiang Y-Q, Ng CH, et al. Extract of ginkgo biloba for tardive dyskinesia: meta-analysis of randomized controlled trials. Pharmacopsychiatry. 2016;49(3):107-111. PubMed CrossRef
43. Pappa S, Tsouli S, Apostolou G, et al. Effects of amantadine on tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Clin Neuropharmacol. 2010;33(6):271-275. PubMed CrossRef
44. Angus S, Sugars J, Boltezar R, et al. A controlled trial of amantadine hydrochloride and neuroleptics in the treatment of tardive dyskinesia. J Clin Psychopharmacol. 1997;17(2):88-91. PubMed CrossRef
45. Leung JG, Breden EL. Tetrabenazine for the treatment of tardive dyskinesia. Ann Pharmacother. 2011;45(4):525-531. PubMed CrossRef
46. Bernstein AI, Stout KA, Miller GW. The vesicular monoamine transporter 2: an underexplored pharmacological target. Neurochem Int. 2014;73:89-97. PubMed CrossRef
47. Kazamatsuri H, Chien C, Cole JO. Treatment of tardive dyskinesia, I: clinical efficacy of a dopamine-depleting agent, tetrabenazine. Arch Gen Psychiatry. 1972;27(1):95-99. PubMed CrossRef
48. Kazamatsuri H, Chien CP, Cole JO. Long-term treatment of tardive dyskinesia with haloperidol and tetrabenazine. Am J Psychiatry. 1973;130(4):479-483. PubMed
49. Ondo WG, Hanna PA, Jankovic J. Tetrabenazine treatment for tardive dyskinesia: assessment by randomized videotape protocol. Am J Psychiatry. 1999;156(8):1279-1281. PubMed
50. Shen V, Clarence-Smith K, Hunter C, et al. Safety and efficacy of tetrabenazine and use of concomitant medications during long-term, open-label treatment of chorea associated with Huntington’s and other diseases. Tremor Other Hyperkinet Mov (N Y). 2013;3. PubMed
51. Xenazine (tetrabenazine): highlights of prescribing information. June 2015. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/021894s010lbl.pdf. Accessed June 16, 2017.
52. Müller T. Valbenazine granted breakthrough drug status for treating tardive dyskinesia. Expert Opin Investig Drugs. 2015;24(6):737-742. PubMed CrossRef
53. Meyer JM. Forgotten but not gone: new developments in the understanding and treatment of tardive dyskinesia. CNS Spectr. 2016;21(S1):13-24. PubMed CrossRef
54. Jankovic J. Dopamine depleters in the treatment of hyperkinetic movement disorders. Expert Opin Pharmacother. 2016;17(18):2461-2470. PubMed CrossRef
55. Ingrezza (valbenazine): highlights of prescribing information. April 2017. http://ingrezza.com/HCP/PI. Accessed September 8, 2017.
56. Austedo (deutetrabenazine): highlights of prescribing information. August 2017. https://www.austedo.com/pi. Accessed September 8, 2017.
57. Meyer JM. Valbenazine for tardive dyskinesia. Curr Psychiatr. 2017;16(5):40-46.
58. O’ Brien CF, Jimenez R, Hauser RA, et al. NBI-98854, a selective monoamine transport inhibitor for the treatment of tardive dyskinesia: a randomized, double-blind, placebo-controlled study. Mov Disord. 2015;30(12):1681-1687. PubMed CrossRef
59. Hauser RA, Factor SA, Marder SR, et al. Kinect 3: a phase 3 randomized, double-blind, placebo-controlled trial of valbenazine for tardive dyskinesia. Am J Psychiatry. 2017;174(5):476-484. PubMed CrossRef
60. Citrome L. Valbenazine for tardive dyskinesia: a systematic review of the efficacy and safety profile for this newly approved novel medication: what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2017;71(7):e12964. PubMed CrossRef
61. Fernandez HH, Factor SA, Hauser RA, et al. Randomized controlled trial of deutetrabenazine for tardive dyskinesia: The ARM-TD study. Neurology. 2017;88(21):2003-2010. PubMed CrossRef
62. Anderson KE, Stamler D, Davis MD, et al. Deutetrabenazine for treatment of involuntary movements in patients with tardive dyskinesia (AIM-TD): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Psychiatry. 2017;4(8):595-604. PubMed CrossRef


![]() Save
Save
![]() Share
Share
![]() Cite
Cite
Advertisement
GAM ID: sidebar-top