
American Society of Clinical Psychopharmacology Corner
J Craig Nelson M.D., Editor
Genetic Studies of Drug Response and Side Effects in the STAR*D Study, Part 1
The Sequenced Treatment Alternatives to Relieve Depression (STAR*D) study was a large clinical trial designed to address the question of the best treatment strategy once a standard first-line pharmaceutical treatment failed for a patient with major depressive disorder (MDD). For STAR*D, the initial treatment for all subjects was citalopram. The trial enrolled over 4,000 subjects, and 1,953 donated DNA for genetic analyses. The key clinical results of this study have been published and reviewed.1
In a series of ancillary studies to STAR*D, several research groups have analyzed the available DNA samples and the corresponding outcome measures from the STAR*D trial in hopes of obtaining insight into the genetic susceptibility to treatment response, the mechanism of antidepressant action, and a number of other research interests. This brief 2-part review highlights the results related to the genetic findings of STAR*D, with focus on the phenotype of remission and adverse side effects including suicide and sexual dysfunction. The well-known serotonin transporter gene will be covered in the first part, and other candidate gene studies, studies of side effects, and future directions for genetic research in the STAR*D sample will be addressed in the second part.
Genetic Association Studies
For all of the studies described below, several principles apply that may be useful to review. Genetic association studies seek to determine if the frequency of an allele at a genetic locus is correlated with the phenotype of interest. If the allele is enriched in cases (those with the phenotype) compared to control subjects at a level that is not likely due to chance, it is considered a risk allele. If the allele under study is enriched in controls compared to cases, it is considered a "protective" allele in that its presence reduces the risk of having the phenotype of interest. For most recent genetic association studies, the loci being studied are single nucleotide polymorphisms (SNPs), which are the most common form of genetic variation in the human genome and consist of having one of (usually) 2 alternate states at a particular base in the genome. The studies reviewed in this article primarily focus on SNPs that do not necessarily lead to a functional alteration in any gene product—they are simply representing (or "tagging") genetic variation in or around genes of interest. In general, the studies described rely on a dichotomous definition of antidepressant response. For example, most studies utilized a remission phenotype in which subjects categorized as remitters had a final score of ≤ 5 on a self-rated or clinician-rated instrument called the Quick Inventory of Depressive Symptomatology (QIDS)2 after a specified minimal treatment duration (eg, 6 weeks). Nonremitters would be categorized as those with scores > 5. Challenges to the data analysis present in the STAR*D sample will be discussed in part 2.
The Serotonin Transporter
The gene most studied in reference to antidepressant response is the serotonin transporter gene (SLC6A4). The gene product (SERT) is responsible for trafficking serotonin back into the presynaptic neuron and thus ending the serotonin signaling cycle. Selective serotonin reuptake inhibitors (SSRIs) affect the serotonin system by blocking the reuptake of serotonin back into the neuron, by interacting with the serotonin transporter and resulting in more serotonin in the synapse. The mechanism by which this occurs is still unclear. The 43 base pair promoter insertion/deletion in SLC6A4 (serotonin transporter linked polymorphic region: 5-HTTLPR) was shown to have differential effects on activity; the long (L; inserted) allele is more transcriptionally active than the short (S; deleted) allele.3 Some studies question whether this is relevant to human brain tissue.4 Altering serotonin levels, typically by use of SSRIs, is speculated as the proximal mechanism underlying therapeutic alleviation of some MDD symptoms. It would be fitting to have DNA variation in this gene associated with antidepressant response, given these biologic considerations. A number of small studies suggest that this might be the case.5 However, given the data to date in STAR*D, this appears not to be the case.
Figure 1. SLC6A4 (serotonin transporter gene)
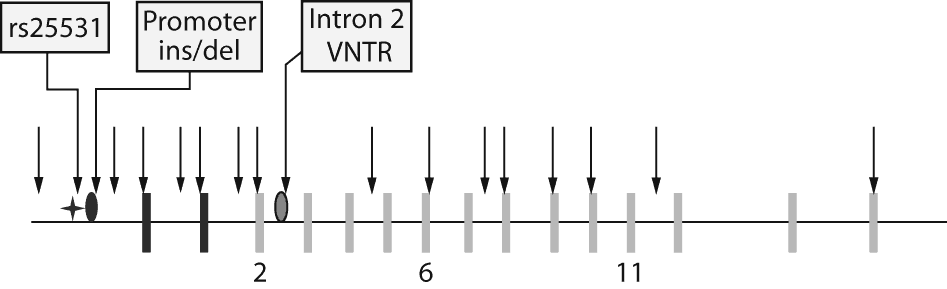
Black rectangles are non-coding exons, and gray rectangles are coding exons. Exons 2, 6, and 11 are indicated. Polymorphisms analyzed by STAR*D researchers are indicated by arrows. The polymorphisms of interest are the rs25531 single nucleotide polymorphism (SNP) (analyzed by Mrazek et al9, Kraft et al,7 and Hu et al8), the promoter insertion/deletion (analyzed by Mrazek et al9, Kraft et al,7 and Hu et al8), and the intron 2 variable number tandem repeat (VNTR) (analyzed by Mrazek et al9). The tally of polymorphisms analyzed, by study, is as follows: Mrazek et al9: 19 SNPs, promoter ins/del, intron 2 VNTR; Kraft et al7: 10 SNPs, promoter ins/del; McMahon et al6: 5 SNPs; Hu et al8: 1 SNP, promoter ins/del.
Click figure to enlarge
SLC6A4 and Remission
Four research groups have addressed the association between genetic variation and MDD remission in response to treatment with citalopram using the STAR*D sample.6-9 Several points should be noted from these studies as a whole: (1) when all studies are considered together, the gene is well covered in terms of genomic coverage; (2) there is an overall lack of association between this gene and citalopram remission; and (3) these studies have their differences, which quite likely contribute to their discordant findings (see discussion below). Two studies7,9 performed novel SNP detection through direct DNA sequencing, which resulted in wider coverage than the promoter area of the gene (Figure 1). When these new SNPs were tested for association with citalopram remission, no association was detected. McMahon et al6 studied 5 SNPs in the SLC6A4 gene and found no association with remission. None of the studies found association between the SLC6A4 variants and remission in the African American subset of STAR*D. This may be due to dramatically reduced statistical power for detecting association in the small African American segment of the sample; thus, a conclusive comment cannot be made until a much larger African American sample is analyzed.
Three groups7-9 assessed association of 2 polymorphisms, the promoter ins/del and rs25531 A/G SNP, with remission status. This latter variant has gained interest due to its potential functional effect on gene function in biochemical and cellular assays. One group utilized a composite "triallelic" genotype of these 2 variants,8 while the other two groups examined haplotypes of these 2 variants in exploratory analyses.7,9 These studies show some variation in terms of phenotype definition and ethnicities analyzed. Similarities include requiring a 6-week participation in the study and no adjustment for ethnicity aside from stratified analyses for each self-reported ethnic group. The one study9 that did find evidence of association between remission and 5-HTTLPR found it among non-Hispanic Caucasians only. The same study found associations between remission and a haplotype of 5-HTTLPR, rs25531, and a variable number tandem repeat (VNTR) in intron 2. The main finding was association between the intron 2 VNTR and remission: non-Hispanic Caucasians without the 12/12 genotype are more likely to achieve remission (P = .02, Pcorrected = .07). None of the studies found associations between remission and the rs25531 A/G SNP alone in any of the STAR*D populations analyzed.
Table 1. Comparison of 3 Studies Investigating the Association Between Remission and 2 Polymorphisms in the SLC6A4 Gene: Promoter ins/del and rs25531a

aMcMahon et al6 not included, since it did not include 5-HTTLPR or rs25531.
bAs determined from global rating of compliance.
cNumber determined from data provided in Table 4 of Mrazek et al9 for promoter insertion/deletion. Numbers are similar for rs25531.
Abbreviations: QIDS-C16 = Quick Inventory of Depressive Symptomatology-Clinician Rated, QIDS-SR = Quick Inventory of Depressive Symptomatology-Self Rated, SNP = single nucleotide polymorphism.
Click figure to enlarge
What underlies the differences between studies that use the same study samples and same polymorphisms? Potential reasons for discordance in results are outlined in Table 1 and include (1) differences in phenotypic definition; (2) ethnic composition of sample (eg, consideration of Hispanic background, or samples analyzed together or separately); and (3) choice of assessment tool (clinician- vs self-rated QIDS). The correlation between remission definitions based on the self-report QIDS and the clinician-report QIDS is quite high (r2 = 0.88); therefore, the assessment tool used to define remission status does not add significantly to the discrepant findings between the studies. It is reassuring to see that even when phenotype definitions are quite disparate, the results are still similar, as in the case of Kraft et al7 and Hu et al.8 One reason the Kraft et al7 and Mrazek et al9 study results are different is due to the ethnic classification; Mrazek et al considered Hispanic background among Caucasians, and Kraft et al did not. Hu et al8 examined Hispanic ethnicity on the same polymorphisms in question and found similar results to Kraft et al,7 concluding that the ethnic stratification was not a likely factor for differential results.
The Mrazek et al9 study did have a larger group of nonremitters compared to the Kraft et al7 and Hu et al8 studies, both of which removed subjects who may have shown a clinical response, but not enough to attain remission. In fact, Mrazek et al is the only study of the group that had more nonremitters than remitters in their analysis.
These subtle phenotype definition differences quite likely contribute to the difference in study results. In aggregate, these results show that SLC6A4, while an obvious candidate for a genetic connection to SSRI response, is not associated with any efficacy phenotype in a large North American sample of individuals with MDD that is most likely to resemble patients seen in typical psychiatric practice. This observation is consistent with recent findings from a sample of 795 subjects from 8 European countries (Genome Based Therapeutic Drugs for Depression [GENDEP]), treated with either escitalopram or nortriptyline, in which 5-HTTLPR was associated with a best fit for response in an interaction model with a recessive mode of inheritance (ie, S/S vs S/L + L/L) in males treated with escitalopram (P = .027), while the intron 2 VNTR was not associated with any response phenotype.10 Interestingly, an association (P = .049) was reported for a SNP 2,500 base pairs downstream of the 5-HTTLPR in the GENDEP sample, although this same SNP was not associated with response (P = .83) in Caucasians (n = 1,308) in the larger STAR*D sample.7 In the next part of this review, we will review the role that other candidate genes play in response phenotypes in STAR*D.
Author affiliations: Department of Psychiatry and Institute for Human Genetics, University of California, San Francisco. Financial disclosure: None reported. Funding/support: None reported. Corresponding author: Steven P. Hamilton, MD, PhD, Department of Psychiatry, University of California, San Francisco, Box NGL, 401 Parnassus Avenue, San Francisco, CA 94143-0984 ([email protected]).
References
1. Warden D, Rush AJ, Trivedi MH, et al. Curr Psychiatry Rep. 2007;9(6):449-459. PubMed doi:10.1007/s11920-007-0061-3
2. Rush AJ, Trivedi MH, Ibrahim HM, et al. Biol Psychiatry. 2003;54(5):573-583. PubMed doi:10.1016/S0006-3223(02)01866-8
3. Lesch K-P, Bengel D, Heils A, et al. Science. 1996;274(5292):1527-1531. PubMed doi:10.1126/science.274.5292.1527
4. Lim JE, Papp A, Pinsonneault J, et al. Mol Psychiatry. 2006;11(7):649-662. PubMed doi:10.1038/sj.mp.4001797
5. Kato M, Serretti A. (published online ahead of print Nov 4, 2008) Mol Psychiatry. 10.1038/mp.2008.116.
6. McMahon FJ, Buervenich S, Charney D, et al. Am J Hum Genet. 2006;78(5):804-814. PubMed doi:10.1086/503820
7. Kraft JB, Peters EJ, Slager SL, et al. Biol Psychiatry. 2007;61(6):734-742. PubMed doi:10.1016/j.biopsych.2006.07.017
8. Hu XZ, Rush AJ, Charney D, et al. Arch Gen Psychiatry. 2007;64(7):783-792. PubMed doi:10.1001/archpsyc.64.7.783
9. Mrazek DA, Rush AJ, Biernacka JM, et al. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(3):341-351. PubMed doi:10.1002/ajmg.b.30816
10. Huezo-Diaz P, Uher R, Smith R, et al. Br J Psychiatry. 2009;195(1):30-38. PubMed doi:10.1192/bjp.bp.108.062521
ASCP Corner offerings are not peer reviewed by the Journal but are peer reviewed by the ASCP. The information contained herein represents the opinion of the author.
Visit the Society Web site at www.ascpp.org
Enjoy free PDF downloads as part of your membership!
![]() Save
Save
![]() Share
Share
![]() Cite
Cite
Advertisement
GAM ID: sidebar-top




{kind=link}
{kind=link}