Article Abstract
The growing number of antipsychotic medications available to treat schizophrenia increases treatment options for patients, but makes clinical decision-making more complex. This article is based on a roundtable discussion by 4 experts on schizophrenia held on November 1, 2011. The goal of this project was to illustrate how to interpret findings from controlled pivotal antipsychotic trials and translate these data into useful information for managing specific patients in clinical settings. The Program to Evaluate the Antipsychotic Response to Lurasidone (PEARL 2) trial was considered relevant for discussion because it was a well-designed registration study that included an active control. Because lurasidone is the most recently approved antipsychotic, clinicians are also likely to be less familiar with this agent. Discussion of the PEARL 2 study served as a starting point for a broader discussion of how to interpret data on newly developed antipsychotic agents for the treatment of schizophrenia. The participants discussed acute and longer term extension findings from the PEARL 2 study in the context of other data on lurasidone, as well as findings concerning other antipsychotics, including the other recently approved agents, asenapine and iloperidone. The panel also provided general guidance for clinicians on how to incorporate newly approved antipsychotics into their clinical practice. Given the relatively modest efficacy differences among available first-line antipsychotics at the patient group level, the panel concluded that clinicians need to focus on safety issues, taking into account patient preferences and past experiences, when selecting the most appropriate antipsychotic for a given patient. They recommended using lower risk agents first, to achieve response with safer agents in as many patients as possible, before moving on to higher risk agents. This paradigm is important because the goal is not simply to achieve acute treatment response, but to maintain that response on a long-term basis with agents that patients can tolerate and that do not significantly increase cardiovascular risk.
ABSTRACT
The growing number of antipsychotic medications available to treat schizophrenia increases treatment options for patients, but makes clinical decision-making more complex. This article is based on a roundtable discussion by 4 experts on schizophrenia held on November 1, 2011. The goal of this project was to illustrate how to interpret findings from controlled pivotal antipsychotic trials and translate these data into useful information for managing specific patients in clinical settings. The Program to Evaluate the Antipsychotic Response to Lurasidone (PEARL 2) trial was considered relevant for discussion because it was a well-designed registration study that included an active control. Because lurasidone is the most recently approved antipsychotic, clinicians are also likely to be less familiar with this agent. Discussion of the PEARL 2 study served as a starting point for a broader discussion of how to interpret data on newly developed antipsychotic agents for the treatment of schizophrenia. The participants discussed acute and longer term extension findings from the PEARL 2 study in the context of other data on lurasidone, as well as findings concerning other antipsychotics, including the other recently approved agents, asenapine and iloperidone. The panel also provided general guidance for clinicians on how to incorporate newly approved antipsychotics into their clinical practice. Given the relatively modest efficacy differences among available first-line antipsychotics at the patient group level, the panel concluded that clinicians need to focus on safety issues, taking into account patient preferences and past experiences, when selecting the most appropriate antipsychotic for a given patient. They recommended using lower risk agents first, to achieve response with safer agents in as many patients as possible, before moving on to higher risk agents. This paradigm is important because the goal is not simply to achieve acute treatment response, but to maintain that response on a long-term basis with agents that patients can tolerate and that do not significantly increase cardiovascular risk.
J Clin Psychiatry InfoPack 2012;5(August):1–12
doi:10.4088/JCP.11078IP1
© Copyright 2012 Physicians Postgraduate Press, Inc.
Faculty
Christoph U. Correll, MD, Chair
Medical Director, Recognition and Prevention Program (RAP), The Zucker Hillside Hospital, Glen Oaks; Associate Professor of Psychiatry, Hofstra North Shore-LIJ School of Medicine, Hempstead; and Adjunct Professor of Psychiatry, Albert Einstein College of Medicine, New York, New York
John Lauriello, MD
Chair and Professor, Department of Psychiatry, University of Missouri and Medical Director, University of Missouri Psychiatric Center, Columbia
Ashok Malla, MD
Professor and Canada Research Chair in Early Psychosis, Department of Psychiatry, McGill University and Director, Prevention and Early Intervention Program for Psychoses, Douglas Institute, Montreal, Quebec, Canada
Jonathan M. Meyer, MD
Assistant Clinical Professor of Psychiatry, Department of Psychiatry, University of California at San Diego and Associate Clinical Professor of Psychiatry, Loma Linda University, Loma Linda, California
Faculty Disclosure
In the spirit of full disclosure, the faculty were asked to complete a statement regarding all relevant personal and financial relationships between themselves or their spouse/partner and any commercial interest. Faculty financial disclosures are as follows:
Dr Correll is a consultant for Alexza, AstraZeneca, Biotis, Bristol-Myers Squibb, Desitin, Eli Lilly, IntraCellular Therapies, Lundbeck, MedAvante, Pfizer, Otsuka, Takeda, and Teva; has received grant/research support from Bristol-Myers Squibb, Otsuka, Janssen, Johnson & Johnson, the National Institute of Mental Health, NARSAD, and the Feinstein Institute for Medical Research; has received honoraria from Bristol-Myers Squibb, Cephalon, GlaxoSmithKline, Janssen, Johnson & Johnson, MedScape, Otsuka, ProPhase, Takeda, and Teva; is a member of the speakers/advisory boards for Actelion, Alexza, AstraZeneca, Bristol-Myers Squibb, IntraCellular Therapies, MedAvante, Merck, Novartis, Otsuka, Pfizer, and Sunovion; and is a stock shareholder in Alexza.
Dr Lauriello is a consultant for Janssen, has received grant/research support from Sunovion, and is a member of the advisory board for Otsuka.
Dr Malla has received grant/research support from Janssen-Ortho (Canada), Bristol-Myers Squibb, Pfizer, and AstraZeneca;
has received honoraria from Janssen-Ortho, Bristol-Myers Squibb, and Pfizer; and is a member of the speakers/advisory boards for Sunovion, Lundbeck, Pfizer, and Bristol-Myers Squibb.
Dr Meyer is a member of the speakers/advisory boards for Arbor Scientia, AstraZeneca, Bristol-Myers Squibb, Janssen, Neuroscience Education Institute, Novartis, Merck, Pfizer, and Sunovion.
Review Process
The faculty for this InfoPack discussed the content in a peer-review planning teleconference, the chair and faculty reviewed the InfoPack for accuracy, and a member of the Journal Editorial Board who is without conflict of interest reviewed the InfoPack to determine whether the material is evidence-based and objective.
Acknowledgment
This InfoPack is based on a roundtable discussion by 4 experts on the treatment of schizophrenia held November 1, 2011. This evidence-based, peer-reviewed InfoPack was prepared by Healthcare Global Village, Inc. Financial support for preparation and dissemination of this InfoPack was provided by Sunovion Pharmaceuticals, Inc. The authors acknowledge Ruth Ross, Project Manager, Healthcare Global Village, Inc., for project management and editorial assistance in developing the manuscript. The opinions expressed herein are those of the authors and do not necessarily reflect the views of Healthcare Global Village, Inc., the publisher, the American Society of Clinical Psychopharmacology, or the commercial supporter.
This article is based on a roundtable discussion by 4 experts on the treatment of schizophrenia held November 1, 2011. The discussion was chaired by Christoph U. Correll, MD; the other panel members were John Lauriello, MD, Ashok Malla, MD, and Jonathan M. Meyer, MD. The panel discussed the results of the Program to Evaluate the Antipsychotic Response to Lurasidone (PEARL 2) study in detail,1,2 to illustrate how to interpret findings from controlled pivotal antipsychotic trials. The panel considered these results both in the context of other published data on lurasidone and, finally, and more briefly, in the context of findings concerning other antipsychotic medications, including the other recently approved agents, iloperidone and asenapine. The goal of this project was to help clinicians better understand how to translate data from such pivotal antipsychotic trials into useful information for managing specific patients in general clinical settings.
The PEARL 2 acute clinical trial was considered a relevant study on which to focus because it was a well-designed registration study that included an active control. Because lurasidone is the most recently approved antipsychotic, clinicians are also likely to be less familiar with this agent. In addition, data from other studies of lurasidone, including a 6-month extension of the acute PEARL 2 study, were available to put the acute findings into context.2–6 Thus, the panel considered the PEARL 2 study a suitable starting point for a broader discussion of issues involved in interpreting data from short-term, regulatory approval studies of antipsychotic agents.7 These issues include study design, choice of active comparator, dosing and titration schedule of the study drug and the comparator, efficacy and tolerability outcomes, and generalizability of findings to clinical settings and longer term use.
As part of the discussions, the panel considered the following questions:
- What are key unmet needs in terms of available antipsychotics?
- What key outcomes should be considered in evaluating findings from clinical trials of new antipsychotics?
- What are considerations when evaluating the results of trials that include active controls for assay sensitivity? How do such studies differ from those that use noninferiority designs to compare the efficacy of 2 or more active agents?
The results of the acute phase PEARL 2 study were published in 2011 by Meltzer et al in the American Journal of Psychiatry.
SHORT-TERM EFFICACY FINDINGS FROM THE PEARL 2 STUDY
Dr Correll began the discussion by noting that the availability of more antipsychotic medications to treat schizophrenia has increased options for patients, but that we still lack reliable biomarkers and other response predictors to help individualize patient care. Thus, clinicians currently choose medications for patients with schizophrenia by balancing considerations of efficacy and safety on a case-by-case basis, taking into account patient and family preferences. To strike such a balance, prescribers need to be aware of the clinical trial evidence base concerning medications from which they can choose. Moreover, they need to understand the trial design and patient selection process used in these studies to adequately interpret the findings and apply them to specific patients in their practice.
To inform this process, the panel first reviewed the efficacy findings from the PEARL 2 acute phase and then commented on issues involved in interpreting such efficacy findings, including dose-response relationships, the effect of baseline severity of symptoms, use of active controls, effect sizes, and placebo response rates, with focus on the relationship of these data to symptom improvement.
Key Outcomes
The PEARL 2 study enrolled 478 acutely ill patients with schizophrenia who had been hospitalized for no more than 2 weeks with an acute exacerbation of psychotic symptoms. The patients were randomly assigned to 6 weeks of double-blind treatment with 40 or 120 mg/d of lurasidone, 15 mg/d of olanzapine (active control included to test for assay sensitivity), or placebo (all medications were dosed once daily in the morning).1
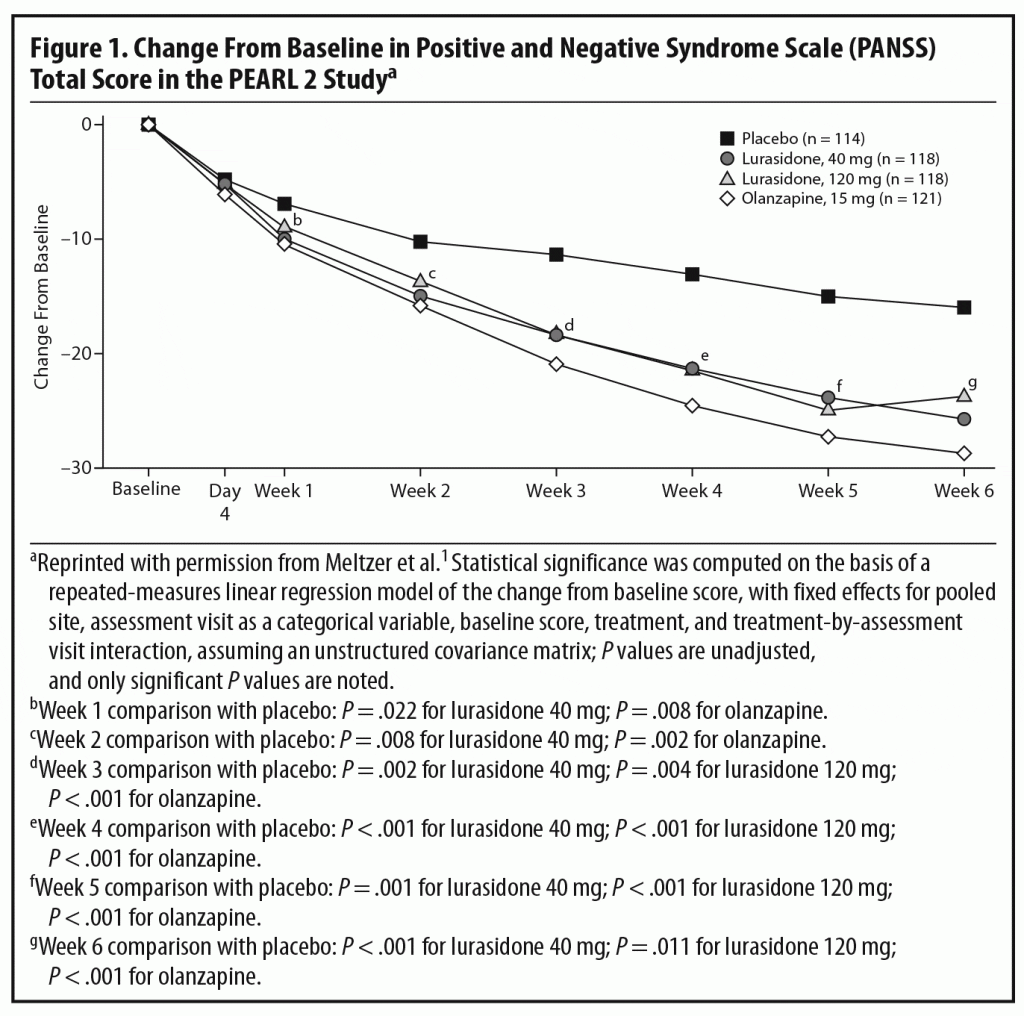
Primary efficacy measure. There was significantly greater improvement from baseline in total scores on the Positive and Negative Syndrome Scale (PANSS)8 at week 6 in patients receiving lurasidone 40 mg/d (−25.7, adjusted P = .002) and 120 mg/d (−23.6, adjusted P = .022) and olanzapine 15 mg/d (−28.7, P < .001) than placebo (−16.0).1
Key secondary efficacy measure. There was significantly greater change from baseline on the Clinical Global Impressions–Severity scale (CGI-S)9 at week 6 with lurasidone 40 mg/d (−1.5, adjusted P = .011) and 120 mg/d (−1.4, adjusted P = .040) and olanzapine (−1.5, P < .001) than with placebo (−1.1).1
Both doses of lurasidone and olanzapine were also associated with significantly greater improvement than placebo at week 6 on the PANSS positive, negative, and general psychopathology subscales and in a post hoc analysis of the modified PANSS cognitive subscale.10 There were no significant differences in improvement on the Montgomery Asberg Depression Rating Scale (MADRS)11 at week 6 between the lurasidone and placebo groups (−3.5, P = .324 and −3.2, P = .571 for lurasidone 40 mg/d and 120 mg/d, respectively, and −2.8 for placebo), while there was a significant difference between results for olanzapine and placebo (−5.0, P = .003). Post hoc analyses found no statistically significant difference between olanzapine and either lurasidone dose nor between the 2 lurasidone doses in mean change in PANSS total scores and CGI-S scores at week 6.1
Dose-Response Relationships and Effect Sizes
In the acute phase of the PEARL 2 study, both 40 and 120 mg/d of lurasidone separated from placebo with no clear dose-response relationship. Olanzapine, the standard comparator, also separated from placebo, confirming the assay sensitivity of the trial (Figure 1). Dr Meyer commented that the moderate effect size (Cohen’s d = 0.43) for mean change in PANSS total score at week 6 for lurasidone 40 mg/d is somewhat on the higher end among more recently approved antipsychotics,1 suggesting that it is an effective dose for patients with schizophrenia. An earlier study by Nakamura et al,3 in which 180 hospitalized patients with an acute exacerbation of schizophrenia were randomized to lurasidone 80 mg/d or placebo for 6 weeks, found an effect size of 0.44 for the 80-mg/d dose, comparable to that for the 40-mg/d dose in this study. The effect size for lurasidone 120 mg/d was somewhat lower at 0.26. Dr Meyer pointed out that this lack of a dose-response relationship may partly reflect the fact that patients receiving 120 mg/d experienced a higher rate of bothersome adverse events (AEs), particularly akathisia, and had a higher rate of discontinuation due to AEs than those receiving the 40-mg/d dose. This higher rate of AEs may have been partly due to administration of the medication in the morning and without titration. Dr Correll noted that, in the later PEARL 3 study,4 in which lurasidone was administered in the evening, rates of akathisia were much lower even with a dose of 160 mg/d (see discussion of safety outcomes below).
Fixed-dose trials, such as the PEARL 2 study, are designed to isolate effects at certain doses. Dr Malla pointed out that, while 120 mg/d might have been a good dose for some patients, it could have been too high for others. In real-world clinical practice, clinicians would generally first try a lower dose, especially in new patients, and then titrate up as needed while monitoring for side effects, whereas, in the PEARL 2 study, patients started immediately at 120 mg/d. In clinical practice, some patients would also stop at a lower dose. In randomized, fixed-dose studies such as the PEARL 2, there is no way to know whether those patients who started immediately on a higher dose would have responded to a lower dose. Similarly, there is no way of knowing if patients who dropped out early could have tolerated and benefited more from a higher dose, if it had been titrated gradually.
Dr Correll noted that the types of patients enrolled in clinical trials may also introduce a selection bias: these patients must be able to read lengthy consent forms and agree to participate; they have fewer comorbid psychiatric and medical conditions than many patients with schizophrenia; and they cannot be involuntarily admitted. Thus, acute randomized, placebo-controlled trials such as this one may actually select for patients who are likely to respond to lower doses than the most severely ill patients who may require higher doses.12 Thus, dose-response relationships may only become detectable in studies that enroll patients who are less responsive to treatment at lower or medium doses. Furthermore, benzodiazepines are also widely used in clinical antipsychotic trials, possibly reducing the need for higher doses in some patients.
The panel agreed that the effects observed in the PEARL 2 study may not be fully representative of what would be seen in actual clinical practice, where doses are adjusted based on illness severity and patient tolerance. Dr Malla stressed the difficulty of resolving such dosing and titration questions based on acute 6-week efficacy studies and the need for Phase 4 effectiveness studies to address such issues. Such studies could include patients who do not respond to the maximum dosing specified in the package insert (derived from fixed-dose studies). In these studies, patients could be randomized to stay on the same dose or be able to go a higher dose, in a manner that is more representative of the decision-making process in clinical care. The panel noted that a similar pattern has occurred with each new antipsychotic agent introduced in recent years, as clinicians have identified the most appropriate dosing for particular patients.13 This highlights the need for clinicians to stay current with information on the most appropriate dosing. For example, the labeling for lurasidone has recently been revised to indicate a maximum dose of 160 mg/d, based on a review of findings from the PEARL 3 study by the US Food and Drug Administration (FDA).4
Baseline Severity
Dr Lauriello pointed out that, in interpreting data from short-term acute phase trials, one must also consider the baseline severity of illness. For example, patients in the PEARL 2 study had fairly high baseline PANSS scores (approximately 96–98). A 25-point drop in PANSS total scores, as seen in this study, is more likely to occur when efficacious treatments are given to patients of this illness severity, while such a dramatic difference compared with placebo might not have been found with less severely ill patients. This means that effects of antipsychotic medications are generally more pronounced in patients with more acute exacerbations of the illness, whereas symptom reductions may be less marked in patients with moderately severe illness.
Active Controls
Dr Meyer observed that, in evaluating a study with an active control arm for assay sensitivity, it is tempting to compare efficacy between the experimental arms (such as between lurasidone and olanzapine in the PEARL 2 study). However, the active control in a placebo-controlled trial is not primarily included to make direct comparisons with the study drug, but for assay sensitivity (ie, to demonstrate that patients included in a study respond as expected when exposed to an active medication with established efficacy). To make direct comparisons of 2 active agents and conclude with a high level of confidence that they are equally efficacious, a noninferiority trial must be done instead, which generally requires a much larger sample size. While it is encouraging that post hoc analyses found no statistically significant separation between lurasidone 40 and 120 mg/d and olanzapine 15 mg/d on the PANSS or the CGI-S scales in the PEARL 2 study, this result should be interpreted cautiously, since this trial was not a noninferiority trial and was quite likely underpowered to make superiority comparisons between the active treatments, rendering it impossible to exclude a potential type II error. The same caution applies to the interpretation of the MADRS results.
Effect Sizes and the Problem of Placebo Response in Clinical Trials of Antipsychotics
All available antipsychotic agents have separated from placebo in terms of efficacy, with efficacy differences among them being relatively small, at least at the group level, with the exception of clozapine for treatment-refractory illness.14–16 Dr Meyer noted that it is difficult to compare antipsychotics in terms of efficacy without head-to-head studies involving the same pool of patients (eg, currently available antipsychotics have been studied at different times across the years and in different populations). In contrast, it is easier to compare safety data across trials, with the caveat that patients’ baseline weight has been increasing in more recent studies.
The effect size for lurasidone of 0.43 discussed above is consistent with those reported for other, recently approved antipsychotics.17–19 However, it is slightly lower than those reported in a 2009 meta-analysis that evaluated 38 randomized controlled trials comparing second-generation antipsychotic drugs (SGAs) with placebo in patients with schizophrenia.14 That meta-analysis, which included 1 study published in the 1980s, 20 studies published in the 1990s, and 17 studies published between 2000 and 2006, found that all of the SGAs were more effective than placebo, with a moderate pooled effect size for overall symptoms of 0.51.
Dr Meyer noted that decreasing effect sizes need to be evaluated in the context of increasing placebo response rates.20 For example, an early placebo-controlled lurasidone trial, which used 3 fixed doses of lurasidone (20 mg, 40 mg, and 80 mg) and haloperidol 10 mg for assay sensitivity, was a failed trial with none of the active treatments demonstrating superiority over placebo.5 This failure of haloperidol 10 mg to separate from placebo illustrates the serious problem of increasing placebo response rates, which has been occurring in studies of all classes of psychiatric medications.
A number of factors may be contributing to increasing placebo response rates. Dr Lauriello noted that, in the United States in recent years, fewer and fewer patients are hospitalized for shorter and shorter periods. Thus, any study involving hospitalization of 2 weeks or longer is essentially providing a kind of milieu treatment that patients may never have received, or not have received in a long time. Dr Malla pointed out that variations in study sites from one country to another may also contribute to this problem. While clinical trials were generally conducted in North America or Europe 10 or 15 years ago, clinical research programs have now been extended to countries where the health care systems and types of patients may be very different from those in developed Western countries. For example, rates of untreated schizophrenia are relatively high in some areas with less widespread psychiatric care12 so that those locations may be enrolling a different type of patient, even though no difference is seen on rating scales. Dr Lauriello noted that effect sizes and placebo response rates at clinical research sites outside North America and Europe initially resembled those in the United States and Europe 15–20 years ago.12 However, as the clinical trials network has grown and become more sophisticated, sites in locations such as Asia, Eastern Europe, and South America have come to more closely resemble those in North America and Western Europe, with rising placebo response rates. This may partly reflect oversampling of the population, which may create “professional patients” who know that, to stay in a study, they need to tolerate the treatment and show improvement or stay in treatment until they can roll over into an open-label extension trial.
Dr Correll noted that, fortunately, despite increasing placebo response rates and lower effect sizes, which make it harder to separate active drugs from placebo, new antipsychotic medications continue to be approved, increasing treatment options for patients with schizophrenia. He pointed out that head-to-head trials and more clinical experience with the newest drugs (eg, asenapine, iloperidone, lurasidone) are needed to inform clinicians about how to effectively use these medications in clinical practice.
SHORT-TERM SAFETY AND TOLERABILITY DATA FROM THE PEARL 2 STUDY
It is important to evaluate the safety profile of each new antipsychotic agent as it is introduced into clinical practice. The panel therefore first briefly discussed the AEs reported in the PEARL 2 study, putting them into the context of AEs with other agents, and then focused on the 4 key safety areas identified by the Schizophrenia Patient Outcomes Research Team (PORT) in their psychopharmacologic treatment recommendations for schizophrenia published in 2010: (1) extrapyramidal side effects (EPS); (2) weight and metabolic effects; (3) prolactin elevation and associated sexual side effects; and (4) QTc prolongation.21
Adverse Events
In selecting an initial antipsychotic agent or deciding to switch medications, clinicians often base their decisions on expected tolerability. Reports of AEs are an important means of evaluating tolerability in clinical trials (Table 1). Dr Correll noted that relatively high rates of AEs are commonly reported in acute antipsychotic trials even with placebo. For example, over 70% of patients in all 4 treatment groups in the PEARL 2 study reported at least 1 AE (placebo, 72%; lurasidone 40 mg/d, 76%; lurasidone 120 mg/d and olanzapine 15 mg/d, 82%), with the majority rated mild to moderate in severity.1 Dr Correll noted that journal articles report the frequency of AE occurrence but often do not incorporate comprehensive data on severity and duration. It is also relevant to look at discontinuation rates due to AEs. These were relatively low in all groups in the PEARL 2 study (lurasidone 40 mg/d, 6.7%; lurasidone 120 mg/d, 11.8%; olanzapine 15 mg/d, 6.5%; placebo, 8.6%). A number of patients in clinical trials are also usually recorded as discontinuing for administrative reasons or withdrawal of consent, and it is possible that some of these discontinuations may actually have been related to AEs or lack of efficacy.
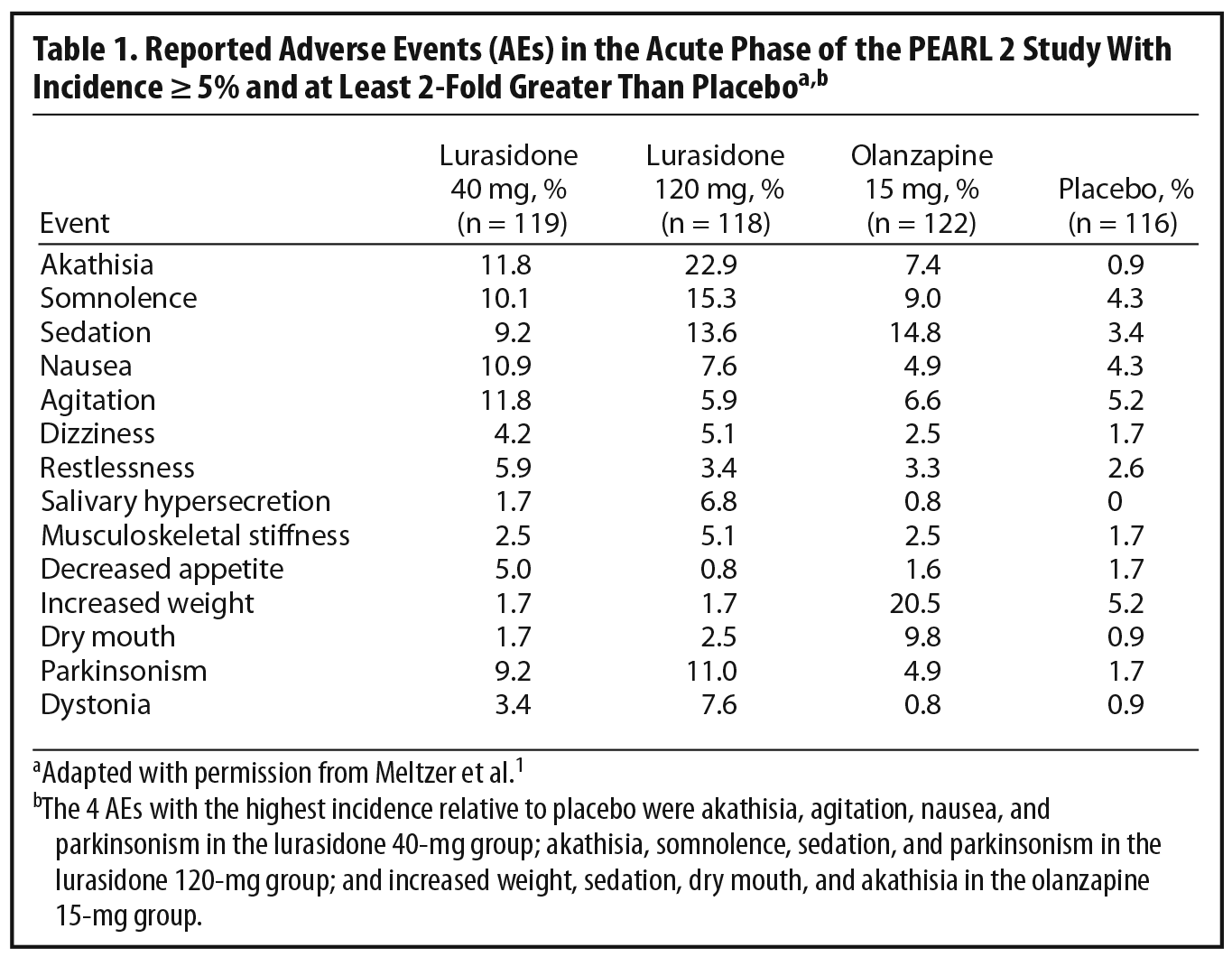
Another important issue in translating clinical trial data into real world practice is whether specific AEs are dose-dependent. In the PEARL 2 acute phase, some AEs that appeared to show a certain amount of dose dependency were akathisia, parkinsonism, dystonia, somnolence, and sedation. The panel noted, however, that the higher rates of akathisia in the lurasidone 120-mg group than in the 40-mg group (22.9% versus 11.8%) might, in part, reflect morning dosing in the PEARL 2 study. This supposition is supported by the fact that, in the PEARL 3 study, in which lurasidone was administered with the evening meal, the incidence of akathisia was 8% in patients receiving 80 mg/d of lurasidone and 7.4% in those receiving 160 mg/d (lurasidone 160 mg/d was also associated with nearly 50% lower rates of parkinsonism in the PEARL 3 study than the 120-mg dose in the PEARL 2 study).4 The panel noted that, in contrast to the PEARL 2 study, the PEARL 3 study did not find any dose dependency in AEs.4
Dr Meyer stressed the importance of considering the potential impact of time of dosing on tolerability in evaluating AEs in clinical trials of antipsychotic agents. He noted that, in clinical practice, clinicians may be reluctant to give an antipsychotic in the morning, because the maximum plasma and brain levels (ie, Tmax), and hence the maximum level of side effects, are likely to occur while the patient is awake. In contrast, if medications are taken in the evening, maximum levels will occur while the patient sleeps. This raises the interesting question if, for example, akathisia rates with other medications, such as first-generation antipsychotics (FGAs) or aripiprazole, would also be lower if the medication were given in the evening.
Dr Meyer noted that discontinuation rates may also shed more light on the importance of specific AEs because patients do, to some extent, “vote with their feet” even in clinical trials. For example, in the PEARL 2 study, only 2 patients (1.7%) dropped out because of akathisia (majority of reports of akathisia were mild or moderate), suggesting that, for many patients, akathisia was not a reason for drop out. The panel noted that akathisia may be a greater contributor to discontinuations in clinical settings, especially among first-episode patients (generally excluded from acute studies), whereas older patients with chronic schizophrenia may be willing to tolerate more side effects in clinical trials.
Dr Malla observed that one outcome missing from short-term acute studies, such as the PEARL 2, is a measure of subjective well-being. Although akathisia was assessed objectively using the Barnes Akathisia Scale (BAS),22 he recommended that future studies of antipsychotics assess patients’ overall sense of well-being, which might reveal differences among antipsychotics not otherwise identified. The panel recommended that future Phase 4 trials place more emphasis on evaluating patient-reported outcomes, drug acceptability to patients, and subjective well-being, which can have a major impact on patients’ decision to continue treatment.
Extrapyramidal Symptoms
Although the risk of EPS is generally much lower with the SGAs than the FGAs,21 EPS can occur with all antipsychotic agents, so that it is important to evaluate results in this area in any study of a new agent. Data on patient-reported EPS were reviewed above. Patients in the PEARL 2 study were also evaluated using the Simpson-Angus Scale (SAS),23 the BAS,22 and the Abnormal Involuntary Movement Scale (AIMS).24 Changes from baseline on the SAS and BAS in the lurasidone 40-mg/d and olanzapine 15-mg/d groups were comparable to those with placebo, while the lurasidone 120-mg group showed significantly greater increases in scores on these scales compared with placebo. The generally low rates of EPS found with lurasidone compared to those with most FGAs are consistent with results from trials of the other SGAs, although it should be noted that rates of EPS are dose dependent and differ among specific SGAs, with risperidone and ziprasidone found to have higher rates than olanzapine, quetiapine, and clozapine.25
Weight and Metabolic Effects
Weight gain and metabolic abnormalities can have an enormous impact on the self-esteem, social functioning, and long-term cardiovascular health of patients with schizophrenia. Findings in the olanzapine group in the PEARL 2 study showing an increase in mean weight (+4.1 kg versus +0.6 kg with placebo) and body mass index (BMI) (+1.4 kg/m2 versus +0.2 kg/m2 with placebo) suggest that patients in this study responded in a way consistent with findings in earlier trials of olanzapine in patients with schizophrenia.21 As expected, given lurasidone’s pharmacodynamic profile,26 changes in weight and BMI with lurasidone were comparable to those with placebo (weight: +1.0 kg with lurasidone versus +0.6 kg with placebo; BMI: +0.4 kg/m2 versus +0.2 kg/m2 with placebo).1 In addition, 34% of those in the olanzapine group had at least a 7% increase in weight over baseline (criterion generally used by the US FDA to indicate significant increase in weight27), compared with 7.6% in the lurasidone 40-mg group, 4.2% in the lurasidone 120-mg group, and 7.0% in the placebo group. Changes at endpoint in cholesterol, triglyceride, high- and low-density lipoprotein (HDL and LDL), and glucose levels were also comparable in the lurasidone and placebo groups, while the olanzapine group had a significant mean increase in cholesterol, triglyceride, LDL, and glucose levels compared with placebo.1 These results place lurasidone in the group of SGAs with low cardiometabolic risk, together with aripiprazole and ziprasidone.28
Dr Correll noted that clinical antipsychotic trials in recent years have involved patient populations with higher mean baseline BMIs, due in part to widespread use of SGAs and increasing levels of overweight and obesity in the general population. Thus, it is possible that newer antipsychotics may show less effect on weight in more recent clinical trials. Thus, it is important to see if patients with no or very limited exposure to antipsychotics will also show minimal weight gain in clinical practice, as it appears that almost all antipsychotics can cause weight gain in young and first-episode patients.28 A large target population for the newest antipsychotics will be overweight or obese patients who have gained weight on other treatments. Phase 4 studies in such patients as well as in adolescents, who are particularly vulnerable to weight gain with antipsychotics,28 will provide a better understanding of how newer agents will perform in these groups and what doses will be most helpful. Moreover, head-to-head trials of lower-risk agents would shed additional light on their relative cardiometabolic profiles.
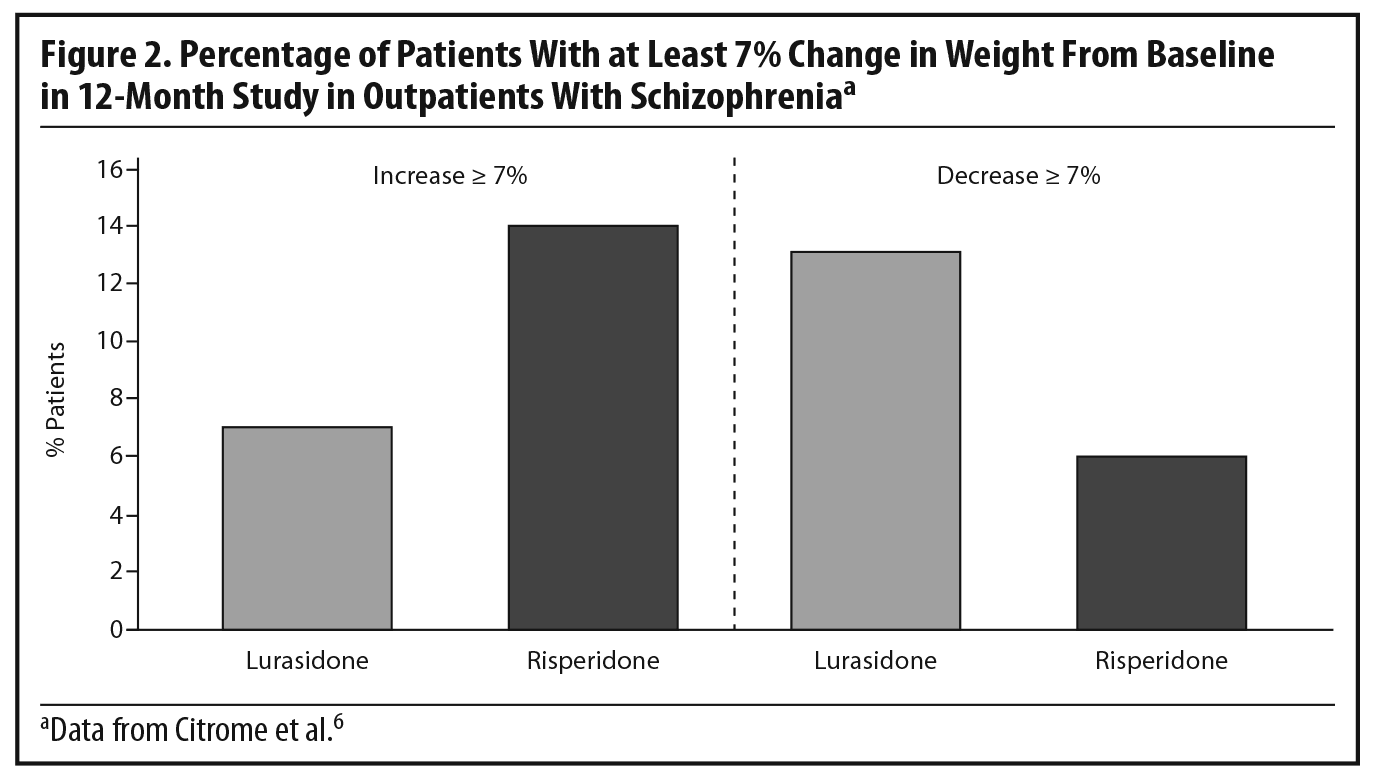
Dr Meyer observed that the short-term weight and metabolic findings for lurasidone are reassuring, noting that, when antipsychotics affect lipid levels, the most notable changes tend to be seen in triglyceride levels.28,29 To put the PEARL 2 results in context, he cited results from a double-blind, randomized, 12-month safety and tolerability study in which clinically stable outpatients with schizophrenia were randomized to flexibly dosed lurasidone 40–120 mg/d (n = 427) or risperidone 2–6 mg/d (n = 202).6 In that study, more patients receiving lurasidone experienced significant weight loss than weight gain (Figure 2), while the opposite occurred with risperidone. Based on observed data at month 12, lurasidone produced a decrease in mean body weight of approximately 0.9 kg at study endpoint compared to an increase of approximately 2.6 kg with risperidone, consistent with findings in other risperidone studies.21
Prolactin Levels
Dr Lauriello noted that, with newer antipsychotics’ improved profiles in terms of EPS, weight, and metabolic effects, elevation in prolactin levels has become a more important concern. Dr Malla commented that increases in prolactin levels and related sexual dysfunction are particularly relevant for younger patients. Young female patients who experience amenorrhea often want to stop their medications, and sexual dysfunction is often particularly problematic for young men with a first episode of schizophrenia, who are more likely to continue to have social and sexual relationships than more chronic patients. Available antipsychotics differ significantly in their propensity to cause clinically significant hyperprolactinemia, with risperidone and paliperidone, followed by the FGAs, being most problematic.21
In the PEARL 2 acute phase, the lurasidone 40-mg and placebo groups showed comparable changes in median prolactin levels (+0.7 and −0.7 ng/mL, P > .05), while statistically significant increases in median prolactin levels compared with placebo occurred in the lurasidone 120-mg (+4.5 ng/mL, P < .001) and olanzapine 15-mg (+3.8 ng/mL, P < .001) groups.1 No patient discontinued treatment because of elevated prolactin and no change in menstrual cycles was observed.1
Dr Correll noted that the changes in prolactin levels with lurasidone 120 mg and olanzapine 15 mg in the PEARL 2 study were comparable and lower than increases reported with risperidone, paliperidone, and the FGAs,30 and, while statistically significant, did not seem clinically relevant. This observation is supported by the 12-month risperidone-controlled study, which found minimal effects on median prolactin levels with lurasidone (−0.5 ng/mL in males and +0.1 ng/mL in females) but significant increases in median prolactin levels with risperidone (+9.6 ng/mL in males and +28.4 ng/mL in females) at study endpoint.6 Dr Lauriello noted that the likelihood of prolactin elevation with lurasidone appears to be small, but outliers always exist and women are more sensitive to these effects than men. Although prolactin levels are not very closely correlated with sexual AEs, it is still important to ask patients about sexual and reproductive dysfunction.
Electrocardiogram (ECG) Abnormalities
Concern has been expressed about QTc prolongation and increased risk of sudden cardiac death with some antipsychotic agents, especially some of the FGAs (eg, thioridazine).31–33 Among the SGAs, ziprasidone, paliperidone, and iloperidone are associated with the greatest increase in QTc intervals, but to date this does not appear to be clinically significant and none of these agents is associated with an increased rate of sudden cardiac death.21,32,33 Nevertheless, changes in QTc intervals are carefully monitored in all regulatory studies of new antipsychotics.
In the acute phase of the PEARL 2 study, no treatment-emergent abnormalities in ECG parameters occurred in any treatment group compared with placebo (mean change in QTcF of +5.1 msec with lurasidone 40 mg, +4.5 msec with lurasidone 120 mg, +4.4 msec with olanzapine, and +3.8 msec with placebo). No QTcF intervals > 450 msec occurred, except for 1 patient with a QTcF > 450 at baseline and throughout the study; no patient had a QTcF > 500 msec or an increase of more than 60 msec in QTcF interval.1
LONGER TERM DATA: FINDINGS FROM THE PEARL 2 EXTENSION STUDY
Patients who successfully completed the 6-week acute phase of the PEARL 2 study were eligible to enter a 6-month open-label extension study (OLE). In this phase, 246 patients were flexibly dosed with 40–120 mg/d of lurasidone for up to 6 months.2 The panel noted that the availability of such longer term data is important in understanding the clinical profile (eg, efficacy, safety, dosing) of new antipsychotic agents. Because the extension phase of the PEARL 2 study involved switching patients from the active control to the study drug, it also provided some preliminary data concerning the effects of switching from olanzapine to lurasidone.
Efficacy
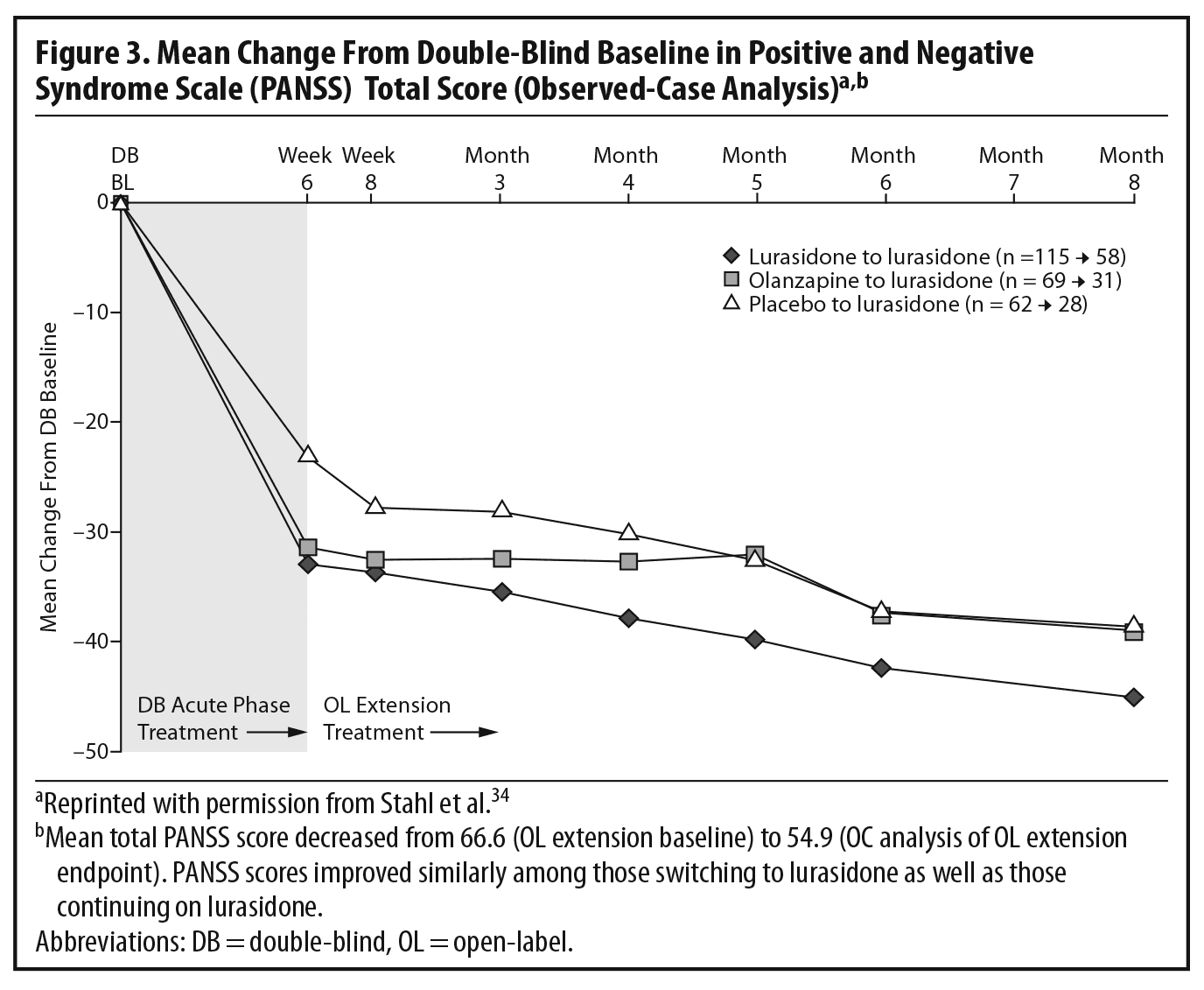
The results of the OLE study showed that the improvement achieved by the end of the acute study (measured by total PANSS score) was maintained over the next 6 months of treatment. Some continued improvement was seen during the extension phase, with small additional improvements seen in all 3 patient subgroups (Figure 3).2,34 The panel noted that, despite being open-label, an extension study like this does, to some extent, simulate real-world clinical care of flexibly dosed maintenance treatment in less acutely ill patients. In addition, these results concerning maintained efficacy are generally consistent with data from open-label extension studies of other approved SGAs.35–39
Dosing
Patients transitioning into the OLE after the acute 6-week trial first underwent a 3-day placebo washout period and were then treated with lurasidone 80 mg/d for 7 days. The dose could then be adjusted between 40 and 120 mg/d up to 4 times during 6 months of treatment (modal lurasidone dose over the extension period was 40 mg in 10%, 80 mg in 63%, and 120 mg in 27% of subjects).2 The panel commented that it was interesting that, when clinicians were free to dose lurasidone as seemed appropriate to them, only 10% received 40 mg/d, while the majority received 80 mg/d. The mean lurasidone dose during the 6-month study period was 86.3 mg/d, suggesting that 80 mg/d may be appropriate for many patients. However, the panel also noted that the mean doses used during the OLE may have been influenced by the starting dose used in the first 7 days of the extension phase. Thus, the OLE may not accurately reflect doses patients would end up receiving if starting lurasidone de novo in clinical practice. Of the 254 patients who entered the OLE (246 patients received at least 1 dose of lurasidone), 113 (44%) completed the 6 months of treatment; 32 (13%) dropped out due to AEs, 17 (7%) dropped out due to insufficient response, and 92 (36%) dropped out for other reasons.2
Safety and Tolerability
The most common AEs (reported in at least 9% of patients) in the 6-month OLE were akathisia (13.0%), insomnia (11.0%), nausea (9.8%), somnolence (9.8%), and parkinsonism (9.3%).2 Only 7.3% reported any AE rated as “severe,” and 13% of patients discontinued treatment during the extension phase due to a treatment-emergent AE. No clinically meaningful changes in vital signs or laboratory findings were observed.2
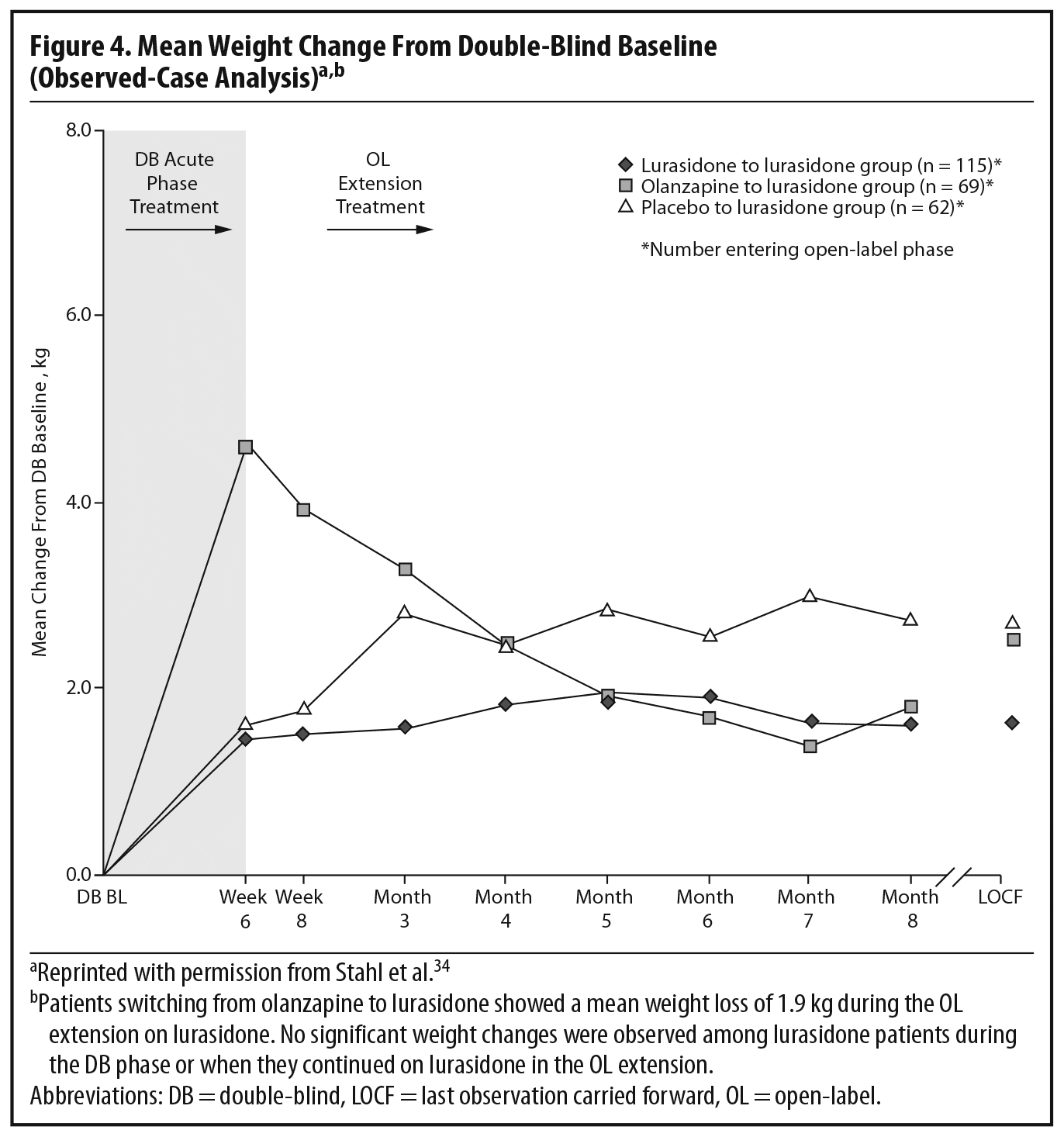
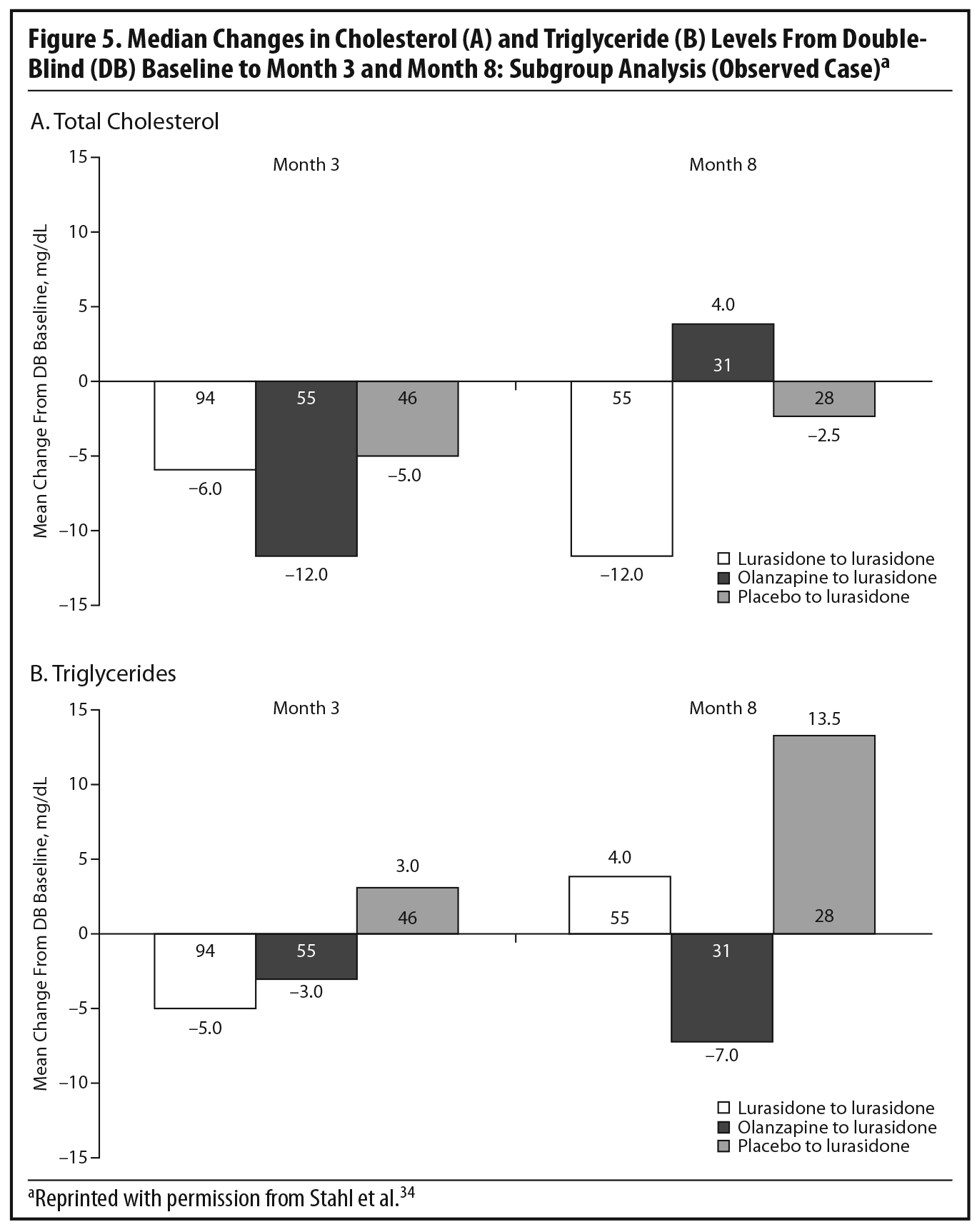
During the OLE, mean body weight (last observation carried forward [LOCF]) was generally stable in patients continuing on lurasidone (+0.1 kg, n = 107) and increased slightly in patients who had switched from placebo to lurasidone (+0.9 kg, n = 55). However, in the patients who had initially been treated with olanzapine 15 mg/d in the acute double-blind phase, there was a mean reduction in weight of 1.9 kg (n = 65) after switching to open-label lurasidone (Figure 4).2,34 This highlights the extent to which the observed cardiometabolic profile of a studied medication can depend on the prior treatment. Similarly, during the OLE, patients who had received olanzapine in the double-blind study (n = 58) experienced larger changes in median lipid levels (−19.0 mg/dL in total cholesterol, −9.5 mg/dL in LDL cholesterol, and −27.5 mg/dL in triglycerides; LOCF) compared with patients who had received lurasidone during the double-blind study (n = 100) (−2.0 mg/dL for total cholesterol, −4.0 mg/dL for LDL cholesterol, and +1.0 mg/dL for triglycerides; LOCF) (Figure 5a and 5b).2
During the OLE, changes in movement disorder signs and symptoms, as measured by the SAS, BARS, and AIMS, were generally absent to mild, and only 4 patients discontinued due to EPS (3 due to akathisia and 1 due to tremor).2 Median prolactin levels decreased over the OLE with lurasidone (−1.3 ng/mL).2 One patient (0.4%) had a ≥ 60 msec increase in QTcF and no patient had a QTcF interval > 500 msec.2 These longer term maintenance results are consistent with findings in OLE programs of other antipsychotics, in that many side effects occur earlier in treatment and that patients who are able to tolerate a medication in the acute phase generally continue tolerating the mediation in the longer term, with the exception of cardiovascular AEs that can increase with time and deserve careful monitoring.
HOW DOES LURASIDONE COMPARE WITH OTHER ANTIPSYCHOTIC AGENTS IN THE TREATMENT OF PATIENTS WITH SCHIZOPHRENIA?
In examining findings from key regulatory studies, such as the PEARL 2 trial, it is important to try to determine how the new agent compares with other available antipsychotics. This information is important because, as Dr Lauriello noted, it can be difficult for clinicians to select the most appropriate agent for a specific patient.
Dr Correll observed that the efficacy of all available antipsychotics, beginning with those introduced over 50 years ago, appears to be mediated by dopamine blockade, so that antipsychotic development has focused largely on refining the side-effect profile of these agents.31 The high-potency FGA haloperidol produced less sedation, weight gain, and constipation than earlier low-potency FGAs (eg, chlorpromazine). The first SGAs were then developed to mitigate the problems with EPS, akathisia, and tardive dyskinesia that were a major drawback with the high-potency FGAs. The focus then shifted to developing agents with less liability for weight gain, sedation, and metabolic abnormalities (problems similar to those seen with low-potency FGAs) that occurred with the first SGAs (eg, clozapine, olanzapine, risperidone, quetiapine), but without paying the price of increased neuromotor side effects. The process of antipsychotic development has thus been one of developing incrementally safer agents that are efficacious for the treatment of schizophrenia.
Given the relatively small differences in efficacy among first-line antipsychotics (with the exception of clozapine for treatment-refractory illness),31 initial prescribing decisions for patients with schizophrenia are often driven by safety concerns. For this reason, the panel recommended reserving drugs associated with more side effects, and of course clozapine, as later options for patients who have failed to respond to a number of different agents and really appear to have treatment-resistant illness.
Dr Lauriello indicated that the main areas of concern since the introduction of the SGAs have been weight gain, metabolic abnormalities, prolactin elevation, and potential cardiotoxicity. He pointed out that asenapine, iloperidone, and lurasidone all appear to have weight and metabolic profiles that are as good or better than other earlier agents with the exception of ziprasidone and aripiprazole.17–19 However, lurasidone seems to stand out by being associated with the least weight gain of the 3, being comparable to placebo, and, at least based on the available data, not appearing to be associated with an increased risk for metabolic syndrome or diabetes and showing no relevant increase in triglyceride or glucose levels.17–19 For example, short-term trials found a mean change in weight of +2.0 kg with iloperidone, +1.1 kg with asenapine, and +0.75 kg or less with lurasidone; longer term trials found small decreases in mean weight from baseline with lurasidone (−0.38 kg at 24 weeks, −0.47 at 36 weeks, and −0.71 at 52 weeks), compared with small increases with asenapine (0.9 kg) and slightly larger increases with iloperidone (4.8 kg).17–19 However, Dr Lauriello also noted that only direct comparisons between these newest agents can confirm this impression, which is based on indirect comparisons. It would also be helpful to evaluate lurasidone in patients who are restarting antipsychotic treatment after a period off medication, who would be at greater risk for weight gain than those who have been chronically treated with antipsychotics.
Dr Correll noted that lurasidone appears to be associated with somewhat more akathisia than the 2 other new agents17–19 (although no direct comparisons with them have been carried out), but that this may be mitigated by administering lurasidone in the evening. Discontinuation rates due to EPS in the lurasidone clinical trials were very low, indicating that this may not be a serious clinical problem for the majority of patients, which is similar to findings concerning the other newer SGAs.
With regard to prolactin levels, of the 3 newest antipsychotics, iloperidone is the agent for which prolactin elevation may be an issue, but elevations are much less than observed with risperidone and paliperidone.18
Neither lurasidone nor asenapine appears to be associated with any ECG abnormalities. QTc prolongation with iloperidone is closer to that observed with ziprasidone,18 although this does not appear to be clinically relevant.40
Lurasidone is administered once a day, while iloperidone and asenapine need to be taken twice a day.17–19 Both asenapine and lurasidone can be started at an effective dose without need for titration. Iloperidone requires dose titration that may take 4 days or more to reach an effective dose. A potential problem with asenapine for some patients is dysgeusia (a distortion of the sense of taste) and some tongue-numbing associated with its sublingual formulation; some patients also find dissolving tablet formulations unwieldy to unwrap and administer, and patients also have to refrain from eating or drinking water for up to 10 minutes to facilitate oral absorption. Lurasidone must be taken with at least 350 kilocalories (kcal) of food, which can be difficult for some patients, particularly those with erratic eating patterns, because, in the fasting state, only about 50% of the prescribed dose is absorbed.41 Dr Meyer suggested explaining to patients that they will get only half of the medication prescribed if lurasidone is taken without food.
To better understand where asenapine, iloperidone, and lurasidone fit among available treatment options, more clinical experience with these agents is needed, as well as Phase 4 studies investigating their use in both more representative and more specific (eg, first-episode, pediatric, geriatric, or morbidly obese) patient populations. Given availability of a number of SGAs in generic formulations, clinicians will be looking for additional benefits in newer agents. More head-to-head studies comparing the newer antipsychotics are needed to provide guidance as to which types of patients are likely to do better on which medications. While none of the newer SGAs appears more effective than earlier agents, the newer SGAs appear to offer some advantages in terms of tolerability and safety. As demonstrated in the CATIE study,42 keeping patients on medication is probably the most effective way to achieve favorable outcomes.
FUTURE RESEARCH DIRECTIONS
The development of new antipsychotics with proven efficacy and improved safety profiles represents progress, but further developments are needed to achieve enhanced outcomes for patients with schizophrenia. For example, a “safer clozapine” is needed that will be effective when first- and second-line dopamine blockers are ineffective.7 Response predictors and biomarkers also need to be developed in order to provide more individualized treatment by predicting which patients might benefit most from which medications, helping to refine treatment selection.43
SELECTING ANTIPSYCHOTIC MEDICATIONS: THE DECISION-MAKING PROCESS
Although it is hoped that research in areas such as genomics, metabolomics, imaging, and genetics will provide clinicians with more accurate tools for medication selection,7,43 clinicians currently need to base treatment decisions on available data. Dr Correll noted that, in selecting the most appropriate medication for a specific patient, clinicians draw on 4 main sources of information (Table 2).44

While one goal of personalized medicine is the development of biomarkers to predict response or AEs, as of 2012 these do not reliably exist for antipsychotics. As clinicians strive to integrate newer agents into their practice, the items listed in Table 2 are the critical elements in determining the most appropriate antipsychotic for a particular patient at a particular moment in the course of treatment. The weighting of the different items in the decision-making process will depend greatly on the stage of illness, prior medication experience and response, psychosocial factors, and patient preference.45 Younger patients early in their course of treatment will be more sensitive to EPS and weight gain,46,47 while more chronic patients may be more concerned about symptomatic relief and be willing to tolerate a certain level of side effect burden. The panel recommended using lower risk agents first, to achieve response with safer agents in as many patients as possible, before moving on to higher risk agents. This paradigm is important because the goal is not simply to achieve acute treatment response, but to maintain that response on a long-term basis with agents that patients can tolerate and that do not significantly increase cardiovascular risk.28,48
Many of these will be individualized decisions, but one important concept that applies to most patients has been gleaned from trials in patients with chronic schizophrenia: early nonresponse after 2 weeks of treatment predicts high likelihood of inadequate response by week 6.31,49–51 The implication is that minimal response after 2 weeks necessitates a rethinking of strategy. The issue may be one of nonadherence, substance use, inadequate dosing, stressors, or the medication itself, but the clinician needs to decide at this stage the next step, and not assume that time alone will solve the problem. Note that this finding does not apply to patients with a first episode of illness, among whom time to initial response varies widely.52,53 Thus, for first-episode patients, a trial of at least 6–8 weeks with gradually increasing dose is generally recommended; target doses for first-episode patients generally turn out to be lower than for patients with more chronic illness.54
The availability of new atypical antipsychotics such as lurasidone presents options for clinicians who want agents with limited potential for metabolic and endocrine AEs,19 as dissatisfaction with side effects is associated with treatment nonadherence.55 The PEARL 2 study provides a reminder that, for any novel medication, clinicians need to develop sophistication in dosing and managing side effects in a manner not necessarily allowable within the study design. For example, more recent lurasidone studies have employed evening dosing with a meal, and available data indicate that doses exceeding those used in the PEARL 2 study (eg, 160 mg) may be better tolerated when given in the evening than 120 mg when administered in the morning.4 Regardless of how well tolerated any new antipsychotic might be, there are clearly patients who will require other options such as clozapine, despite its metabolic liabilities, or a depot antipsychotic in treatment responders for whom nonadherence is the primary barrier to effective symptom management.55 One also cannot minimize the importance of psychoeducation for patients and families, cognitive therapy for symptom management, and psychosocial rehabilitation to extend the gains made from pharmacotherapy.56
CONCLUSION
In this roundtable discussion, the panel examined data from the acute and long-term extension phases of the lurasidone PEARL 2 clinical trial as a starting point for a broader discussion of how to interpret data on newly developed antipsychotics. They also discussed how this new agent compares with other available antipsychotics. The panel focused specifically on lurasidone as an example, because it is the most recently approved agent with which clinicians may have the least clinical experience. The authors hope that this discussion will enrich clinical care by helping clinicians better understand how to translate clinical trial data into clinical practice.
Drug names: aripiprazole (Abilify), asenapine (Saphris), clozapine (Clozaril, FazaClo, and others), haloperidol (Haldol and others), iloperidone (Fanapt), lurasidone (Latuda), olanzapine (Zyprexa), paliperidone (Invega), quetiapine (Seroquel), risperidone (Risperdal and others), ziprasidone (Geodon).
REFERENCES
1. Meltzer HY, Cucchiaro J, Silva R, et al. Lurasidone in the treatment of schizophrenia: a randomized, double-blind, placebo- and olanzapine-controlled study. Am J Psychiatry. 2011;168(9):957–967. PubMed Show Abstract
2. Stahl SM, Cucchiaro J, Simonelli D, et al. Long-term safety and tolerability of lurasidone for patients with schizophrenia: results of a 6-month, open-label extension study. Presented at the 164th Annual Meeting of the American Psychiatric Association; May 2011; Honolulu, HI.
3. Nakamura M, Ogasa M, Guarino J, et al. Lurasidone in the treatment of acute schizophrenia: a double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(6):829–836. PubMed doi:10.4088/JCP.08m04905 Show Abstract
4. Loebel A, Cucchiaro J, Sarma K, et al. Lurasidone in the treatment of acute schizophrenia: results of the double-blind, placebo-controlled, 6-week, PEARL 3 trial. Presented at the 164th Annual Meeting of the American Psychiatric Association; May 2011; Honolulu, HI.
5. Citrome L. Lurasidone for schizophrenia: a review of the efficacy and safety profile for this newly approved second-generation antipsychotic. Int J Clin Pract. 2011;65(2):189–210. PubMed doi:10.1111/j.1742-1241.2010.02587.x Show Abstract
6. Citrome L, Cucchiaro J, Sarma K, et al. Long-term safety and tolerability of lurasidone in schizophrenia: a 12-month, double-blind, active-controlled study. Int Clin Psychopharmacol. 2012;27(3):165–176. PubMed doi:10.1097/YIC.0b013e32835281ef Show Abstract
7. Correll CU. What are we looking for in new antipsychotics? J Clin Psychiatry. 2011;72(suppl 1):9–13. PubMed doi:10.4088/JCP.10075su1.02 Show Abstract
8. Kay SR, Fiszbein A, Opler LA. The Positive and Negative Syndrome Scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13(2):261–276. PubMed Show Abstract
9. Guy W. ECDEU Assessment Manual for Psychopharmacology—Revised (DHHS Publ No. ADM 91-338). Rockville, MD: US Department of Health and Human Services; 1976:218–222.
10. Lindenmayer J-P, Grochowski S, Hyman RB. Five factor model of schizophrenia: replication across samples. Schizophr Res. 1995;14(3):229–234. PubMed doi:10.1016/0920-9964(94)00041-6 Show Abstract
11. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134(4):382–389. PubMed doi:10.1192/bjp.134.4.382 Show Abstract
12. Correll CU, Kishimoto T, Kane JM. Randomized controlled trials in schizophrenia: opportunities, limitations and novel trial designs. Dialogues Clin Neurosci. 2011;13(2):155–172. PubMed Show Abstract
13. Citrome L, Reist C, Palmer L, et al. Dose trends for second-generation antipsychotic treatment of schizophrenia and bipolar disorder. Schizophr Res. 2009;108(1–3):238–244. PubMed doi:10.1016/j.schres.2008.11.017 Show Abstract
14. Leucht S, Arbter D, Engel RR, et al. How effective are second-generation antipsychotic drugs? a meta-analysis of placebo-controlled trials. Mol Psychiatry. 2009;14(4):429–447. PubMed doi:10.1038/sj.mp.4002136 Show Abstract
15. Leucht S, Corves C, Arbter D, et al. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet. 2009;373(9657):31–41. PubMed doi:10.1016/S0140-6736(08)61764-X Show Abstract
16. Leucht S, Komossa K, Rummel-Kluge C, et al. A meta-analysis of head-to-head comparisons of second-generation antipsychotics in the treatment of schizophrenia. Am J Psychiatry. 2009;166(2):152–163. PubMed doi:10.1176/appi.ajp.2008.08030368 Show Abstract
17. Potkin SG. Asenapine: a clinical overview. J Clin Psychiatry. 2011;72(suppl 1):14–18. PubMed doi:10.4088/JCP.10075su1.03 Show Abstract
18. Citrome L. Iloperidone: a clinical overview. J Clin Psychiatry. 2011;72(suppl 1):19–23. PubMed doi:10.4088/JCP.10075su1.04 Show Abstract
19. Kane JM. Lurasidone: a clinical overview. J Clin Psychiatry. 2011;72(suppl 1):24–28. PubMed doi:10.4088/JCP.10075su1.05 Show Abstract
20. Kinon BJ, Potts AJ, Watson SB. Placebo response in clinical trials with schizophrenia patients. Curr Opin Psychiatry. 2011;24(2):107–113. PubMed Show Abstract
21. Buchanan RW, Kreyenbuhl J, Kelly DL, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychopharmacological treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):71–93. PubMed doi:10.1093/schbul/sbp116 Show Abstract
22. Barnes TRE. A rating scale for drug-induced akathisia. Br J Psychiatry. 1989;154(5):672–676. PubMed doi:10.1192/bjp.154.5.672 Show Abstract
23. Simpson GM. Angus JWS. A rating scale for extrapyramidal side effects. Acta Psychiatr Scand suppl. 1970;212:11–19. PubMed doi:10.1111/j.1600-0447.1970.tb02066.x Show Abstract
24. Guy W, ed. ECDEU Assessment Manual for Psychopharmacology—Revised (DHEW Publ No. ADM 76-338). Rockville, MD: US Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, NIMH Psychopharmacology Research Branch, Division of Extramural Research Programs 1976: 534-537.
25. Rummel-Kluge C, Komossa K, Schwarz S, et al. Second-generation antipsychotic drugs and extrapyramidal side effects: a systematic review and meta-analysis of head-to-head comparisons. Schizophr Bull. 2012;38(1):167–177. PubMed doi:10.1093/schbul/sbq042 Show Abstract
26. Ishibashi T, Horisawa T, Tokuda K, et al. Pharmacological profile of lurasidone, a novel antipsychotic agent with potent 5-hydroxytryptamine 7 (5-HT7) and 5-HT1A receptor activity. J Pharmacol Exp Ther. 2010;334(1):171–181. PubMed doi:10.1124/jpet.110.167346 Show Abstract
27. 2012 Physicians’ Desk Reference (PDR). 66th ed. Montvale, NJ: PDR Network; 2012.
28. De Hert M, Detraux J, van Winkel R, et al. Metabolic and cardiovascular adverse effects associated with antipsychotic drugs. Nat Rev Endocrinol. 2012;8(2):114–126. PubMed doi:10.1038/nrendo.2011.156 Show Abstract
29. Meyer JM, Koro CE. The effects of antipsychotic therapy on serum lipids: a comprehensive review. Schizophr Res. 2004;70(1):1–17. PubMed doi:10.1016/j.schres.2004.01.014 Show Abstract
30. Bostwick JR, Guthrie SK, Ellingrod VL. Antipsychotic-induced hyperprolactinemia. Pharmacotherapy. 2009;29(1):64–73. PubMed Show Abstract
31. Kane JM, Correll CU. Past and present progress in the pharmacologic treatment of schizophrenia. J Clin Psychiatry. 2010;71(9):1115–1124. PubMed doi:10.4088/JCP.10r06264yel Show Abstract
32. Glassman AH, Bigger JT Jr. Antipsychotic drugs: prolonged QTc interval, torsade de pointes, and sudden death. Am J Psychiatry. 2001;158(11):1774–1782. PubMed doi:10.1176/appi.ajp.158.11.1774 Show Abstract
33. Ray WA, Chung CP, Murray KT, et al. Atypical antipsychotic drugs and the risk of sudden cardiac death. N Engl J Med. 2009;360(3):225–235. PubMed doi:10.1056/NEJMoa0806994 Show Abstract
34. Stahl SM, Bollu VK, Rajagopalan K, et al. Switching from olanzapine to lurasidone: results from a 6-month open label extension study. Poster Presented at the 165th Annual Meeting of the American Psychiatric Association; May 7, 2012; Philadelphia, PA.
35. Kane JM, Lauriello J, Laska E, et al. Long-term efficacy and safety of iloperidone: results from 3 clinical trials for the treatment of schizophrenia. J Clin Psychopharmacol. 2008;28(suppl 1):S29–S35. PubMed doi:10.1097/JCP.0b013e318169cca7 Show Abstract
36. Kozma CM, Slaton T, Dirani R, et al. Changes in schizophrenia-related hospitalization and ER use among patients receiving paliperidone palmitate: results from a clinical trial with a 52-week open-label extension (OLE). Curr Med Res Opin. 2011;27(8):1603–1611. PubMed doi:10.1185/03007995.2011.595000 Show Abstract
37. Emsley R, Berwaerts J, Eerdekens M, et al. Efficacy and safety of oral paliperidone extended-release tablets in the treatment of acute schizophrenia: pooled data from three 52-week open-label studies. Int Clin Psychopharmacol. 2008;23(6):343–356. PubMed doi:10.1097/YIC.0b013e328314e1f3 Show Abstract
38. Chrzanowski WK, Marcus RN, Torbeyns A, et al. Effectiveness of long-term aripiprazole therapy in patients with acutely relapsing or chronic, stable schizophrenia: a 52-week, open-label comparison with olanzapine. Psychopharmacology (Berl). 2006;189(2):259–266. PubMed doi:10.1007/s00213-006-0564-3 Show Abstract
39. Kasper S, Brecher M, Fitton L, et al. Maintenance of long-term efficacy and safety of quetiapine in the open-label treatment of schizophrenia. Int Clin Psychopharmacol. 2004;19(5):281–289. PubMed doi:10.1097/01.yic.0000138822.62874.fd Show Abstract
40. Strom BL, Eng SM, Faich G, et al. Comparative mortality associated with ziprasidone and olanzapine in real-world use among 18,154 patients with schizophrenia: the Ziprasidone Observational Study of Cardiac Outcomes (ZODIAC). Am J Psychiatry. 2011;168(2):193–201. PubMed doi:10.1176/appi.ajp.2010.08040484 Show Abstract
41. Latuda [prescribing information]. Marlborough, MA: Sunovion Pharmaceuticals. http://www.latuda.com/LatudaPrescribingInformation.pdf. Updated May 2011. Accessed January 15, 2012.
42. Lieberman JA, Stroup TS, McEvoy JP, et al; Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) Investigators. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209–1223. PubMed doi:10.1056/NEJMoa051688 Show Abstract
43. Correll CU, Cañas F, Larmo I, et al. Individualizing antipsychotic treatment selection in schizophrenia: characteristics of empirically derived patient subgroups. Eur Psychiatry. 2011;26(suppl 1):3–16. PubMed doi:10.1016/S0924-9338(11)71709-6 Show Abstract
44. Kane JM, Correll CU. Pharmacologic treatment of schizophrenia. Dialogues Clin Neurosci. 2010;12(3):345–357. PubMed Show Abstract
45. Glick ID, Correll CU, Altamura AC, et al. Mid-term and long-term efficacy and effectiveness of antipsychotic medications for schizophrenia: a data-driven, personalized clinical approach. J Clin Psychiatry. 2011;72(12):1616–1627. PubMed doi:10.4088/JCP.11r06927 Show Abstract
46. Correll CU. Antipsychotic use in children and adolescents: minimizing adverse effects to maximize outcomes. J Am Acad Child Adolesc Psychiatry. 2008;47(1):9–20. PubMed doi:10.1097/chi.0b013e31815b5cb1 Show Abstract
47. Correll CU, Manu P, Olshanskiy V, et al. Cardiometabolic risk of second-generation antipsychotic medications during first-time use in children and adolescents. JAMA. 2009;302(16):1765–1773. PubMed doi:10.1001/jama.2009.1549 Show Abstract
48. Raedler TJ. Cardiovascular aspects of antipsychotics. Curr Opin Psychiatry. 2010;23(6):574–581.
49. Correll CU, Kishimoto T, Nielsen J, et al. Quantifying clinical relevance in the treatment of schizophrenia. Clin Ther. 2011;33(12):B16–B39. PubMed doi:10.1016/j.clinthera.2011.11.016 Show Abstract
50. Leucht S, Busch R, Kissling W, et al. Early prediction of antipsychotic nonresponse among patients with schizophrenia. J Clin Psychiatry. 2007;68(3):352–360. PubMed doi:10.4088/JCP.v68n0301 Show Abstract
51. Leucht S, Shamsi SA, Busch R, et al. Predicting antipsychotic drug response—replication and extension to six weeks in an international olanzapine study. Schizophr Res. 2008;101(1–3):312–319. PubMed doi:10.1016/j.schres.2008.01.018 Show Abstract
52. Emsley R, Rabinowitz J, Medori R. Time course for antipsychotic treatment response in first-episode schizophrenia. Am J Psychiatry. 2006;163(4):743–745. PubMed doi:10.1176/appi.ajp.163.4.743 Show Abstract
53. Gallego JA, Robinson DG, Sevy SM, et al. Time to treatment response in first-episode schizophrenia: should acute treatment trials last several months? J Clin Psychiatry. 2011;72(12):1691–1696. PubMed doi:10.4088/JCP.10m06349 Show Abstract
54. Robinson DG, Woerner MG, Delman HM, et al. Pharmacological treatments for first-episode schizophrenia. Schizophr Bull. 2005;31(3):705–722. PubMed doi:10.1093/schbul/sbi032 Show Abstract
55. Velligan DI, Weiden PJ, Sajatovic M, et al; Expert Consensus Panel on Adherence Problems in Serious and Persistent Mental Illness. The expert consensus guideline series: adherence problems in patients with serious and persistent mental illness. J Clin Psychiatry. 2009;70(suppl 4):1–46, quiz 47–48. PubMed Show Abstract
56. Dixon LB, Dickerson F, Bellack AS, et al; Schizophrenia Patient Outcomes Research Team (PORT). The 2009 schizophrenia PORT psychosocial treatment recommendations and summary statements. Schizophr Bull. 2010;36(1):48–70. PubMed doi:10.1093/schbul/sbp115 Show Abstract
![]() Save
Save
![]() Share
Share
![]() Cite
Cite
Advertisement
GAM ID: sidebar-top