Pregnancy labeling of prescription medications in the US is in the midst of a major transformation. The FDA’s previous system, which used letter ratings to convey drug safety, was simple but led to misunderstandings—both faulty assurances and undue concerns. The new system, established under the Pregnancy and Lactation Labeling Rule, aims for more descriptive and up-to-date explanations of risk as well as context needed for informed decision-making based on available data. In April 2017, a conference titled “Pharmacovigilance, Reproductive Safety, and the Pregnancy and Lactation Labeling Rule” brought together clinicians and researchers, FDA officials, and representatives of the public and industry to discuss a host of questions relating to the new system. This Academic Highlights article summarizes their discussions, which included topics such as how the new system came about and how the new labeling can be used effectively to inform physician-patient conversations about use of medications during pregnancy and ultimately the clinical decisions that follow.’ ‹’ ‹

This Academic Highlights section of The Journal of Clinical Psychiatry presents the highlights of the interactive conference “Pharmacovigilance, Reproductive Safety, and the Pregnancy and Lactation Labeling Rule,” which was held in April 2017.
The course directors were Marlene P. Freeman, MD, and Lee S. Cohen, MD, of Massachusetts General Hospital, Boston, Massachusetts. The faculty were Tiffany Farchione, MD; Lynn Yao, MD; Leyla Sahin, MD; and Lockwood Taylor, PhD, of the US Food and Drug Administration, Silver Spring, Maryland; Krista F. Huybrechts, MS, PhD, of Harvard Medical School, Boston, Massachusetts; Ruta Nonacs, MD, PhD, of Massachusetts General Hospital, Boston, Massachusetts; and Adele C. Viguera, MD, of Massachusetts General Hospital, Boston, Massachusetts, and Cleveland Clinic, Cleveland, Ohio. The program manager was Alexandra Z. Sosinsky, BS, of Massachusetts General Hospital, Boston, Massachusetts.
Potential conflicts of interest: Dr Freeman has received research support from Alkermes, Otsuka, Forest/Actavis, Sunovion, Teva, JayMac, Takeda/Lundbeck, and SAGE Therapeutics; has received payment for medical editing from GOED Omega-3 Newsletter; has been on an Independent Data Monitoring Committee for Johnson & Johnson/Janssen; and works with the MGH Clinical Trials Network and Institute, which has received research funding from multiple pharmaceutical companies. Dr Huybrechts has received research support from Eli Lilly, Pfizer, Boehringer Ingelheim, and GlaxoSmithKline. Dr Nonacs has received royalties from Simon & Schuster. Dr Viguera has received research support from Alkermes, Otsuka, Forest/Actavis, Sunovion, and Teva. Dr Cohen has received research support from Alkermes, Otsuka, Forest/Actavis, Sunovion, Teva, JayMac, and Takeda/Lundbeck. Drs Farchione, Yao, Sahin, and Taylor and Ms Sosinsky report no potential conflicts of interest.
Conference participants
Academia: Catherine Birndorf, MD, Weill Cornell Medical Center | Shari Lusskin, MD, Icahn School of Medicine at Mount Sinai | Lauren Osborne, MD; Jennifer Payne, MD; and Jessica Katznelson, MD, Johns Hopkins | Michele Preminger, MD, Women’s Emotional Wellness Center | Lisa Weinstock, MD, New York Medical College
Government: Caitlin Cusack, MD, MPH, CPHIMS, FHIMSS, Department of Veterans Affairs | Wei Hua, MD, PhD; Judith Zander, MD; Karen Feibus, MD; and Catherine Roca, MD, US Food and Drug Administration | Perdita Taylor-Zapata, MD, National Institutes of Health
Industry: John Affinito, MD, Takeda | Debra Feldman, MPH, and Leslie Williams, DVM, MPH, SAGE Therapeutics | Jehan Marino, PharmD, BCPP, and Robert Pitasi, PharmD, BCPP, MBA, Otsuka Pharmaceutical | Irma Saliu, PharmD, Allergan | Gary Bloomgren, MD, MBA, Alkermes | Judith Kando, PharmD, Shire | Patricia Kauffman, MD, MA, Acadia Pharmaceuticals | Amy O’ Donnell, MD, JD, Johnson & Johnson | Patrick Hu, MD, AstraZeneca | Diana Hughes, MBBS, MSc, Sunovion
Advocacy: Stephen Braddock, MD, and Melissa Tassinari, PhD, Organization of Teratology Information Specialists | Samantha Dowd, BA, BS, Postpartum Support International
MGH Psychiatry Academy staff: Grace Shanks, BA, and Jane Pimental, MPH
The conference from which this report was derived received support from Takeda, Sunovion, and Alkermes in the form of educational grants to the MGH Psychiatry Academy. These funds were also used to support assistance from Christine Kilgore, BA, with the medical writing of the manuscript.
J Clin Psychiatry 2018;79(4):18ah38120
To cite: Freeman MP, Farchione T, Yao L, et al. Psychiatric medications and reproductive safety: scientific and clinical perspectives pertaining to the US FDA Pregnancy and Lactation Labeling Rule. J Clin Psychiatry. 2018;79(4):18ah38120
To share: https://doi.org/10.4088/JCP.18ah38120
© Copyright 2018 Physicians Postgraduate Press, Inc.
Pregnancy labeling of prescription medications is in the midst of a major transformation. Historically, the US Food and Drug Administration (FDA) has had a pregnancy category labeling system that used the letters A, B, C, D, and X to convey reproductive and lactation safety. The system was attractive for its perceived simplicity, but these risk categories led to misunderstandings—both faulty assurances and unduly heightened concerns. The new system, established under the Pregnancy and Lactation Labeling Rule (PLLR), aims for more descriptive and up-to-date explanations of risk as well as context needed for informed decision-making based on available data (https://www.federalregister.gov/a/2014-28241). By June 30, 2018, prescription drugs will no longer have pregnancy letter categories, and by June 30, 2020, all drugs approved since June 2001 will be in the new format.
In April 2017, a 1-day interactive conference titled “Pharmacovigilance, Reproductive Safety, and the Pregnancy and Lactation Labeling Rule” brought together clinicians and researchers, FDA officials, and representatives of the public and industry to discuss a host of questions, including these: How did the new system come about? How can information in the new labeling be most effectively communicated so that it can optimally inform physician-patient conversations about use of medications during pregnancy and ultimately clinical decisions that follow? and Should postmarketing pharmacovigilance change to provide more meaningful data for patients and clinicians?
The meeting was planned collaboratively by representatives of the FDA and faculty at the Ammon-Pinizzotto Center for Women’s Mental Health at Massachusetts General Hospital and directly overseen by the MGH Psychiatry Academy. It was codirected by Marlene P. Freeman, MD, and Lee S. Cohen, MD, both of the MGH Center for Women’s Mental Health. This Academic Highlights section presents a summary of the meeting’s presentations and discussions.
History of Pregnancy Labeling and Development and Adoption of the PLLR
Lynne Yao, MD, and Leyla Sahin, MD, both of the FDA’s Division of Pediatric and Maternal Health in the Office of New Drugs, Center for Drug Evaluation and Research, described the intent and status of the PLLR in separate presentations. Published on December 4, 2014,1 and effective June 30, 2015, the PLLR is intended to provide the prescriber with a descriptive, more meaningful, and more up-to-date statement of the known risks of exposure to medications during pregnancy and lactation.
Evolution of Pregnancy Labeling for Prescription Drugs
Dr Yao opened the meeting by discussing the history of pregnancy labeling for prescription drugs, the status of the PLLR, and implementation challenges. She emphasized that the PLLR is the culmination of more than 25 years of input (Table 1) from researchers, clinicians, patients, and other stakeholders aimed at improving the content of pregnancy information in product labeling and how it is communicated to health care providers.
Pregnancy labeling dates back to 1979, when the FDA issued its first labeling requirements for reproductive safety and instructed manufacturers to assign each product to a pregnancy risk category of A, B, C, D, or X. According to Dr Yao, the tendency of health care providers and the public to misinterpret the letter categories as a grading system became problematic soon after the system was implemented. For example, category C was especially confusing and ambiguous because it covered both drugs for which there was adverse reproduction information from animal studies but no adequate human studies and drugs for which there were no studies at all in either animals or pregnant women.
Concerns about the misinterpretation of the letter category system and calls for change increased in the early 1990s, Dr Yao explained. The Teratology Society recommended to the FDA that it eliminate the pregnancy letter categories altogether and instead provide narrative statements that summarize and interpret available data about teratogenic hazards. In response, in 1994, the FDA established a Pregnancy Labeling Task Force to consider ways of making pregnancy labeling more consistent and informative.
At a subsequent hearing convened in 1997, the public had an opportunity to provide comments on the process and shared various concerns, including that drugs in a particular letter category were incorrectly assumed to carry similar degrees of risk and that categories did not distinguish between data from animals and humans. The FDA was also urged to distinguish between risk information and clinical management information in pregnancy labeling. Dr Yao noted that the majority of drugs, about 65% to 70%, were labeled as Category C by this time.

Click figure to enlarge
“The hearing brought the whisperings and beginnings of what we have today,” Dr Yao said. Between 1979 and 1997, she emphasized, there had been vast improvements in information collection, public attention to issues of reproductive safety and decision-making, and the sophistication of risk communication. In 1999, the FDA published a white paper that outlined potential changes to the format and content of labeling, including replacement of the letter category designations with narrative text and a concise summary of risks. The agency convened a special subcommittee of the Reproductive Health Drugs Advisory Committee, as well as clinician-comprised focus groups, to assist in refining its draft model labeling. In 2008, the FDA published its proposed rule for Pregnancy and Lactation Labeling.

- The previous pregnancy labeling system using letter categories for prescription medications is no longer in effect. The new labeling provides descriptive reproductive safety data, prioritizes human data over animal data, and includes context for risks.
- The risk/benefit assessment for the use of prescription medication in pregnancy includes the available data regarding demonstrated risks, as well as the risks of the untreated disorder if medication is discontinued.
- Patients and health care providers seek information about the safety of medications during pregnancy and breastfeeding from a number of sources, and education and good communication around these topics are essential.
The proposed rule removed the letter categories and replaced them with a new format. The proposed rule also required that information on pregnancy exposure registries be highlighted in the labeling (in hopes of encouraging participation) and that labeling include a standard required background risk statement, Dr Yao explained.
She summarized the major changes between this proposed rule and the final rule published in 2014. These included replacement of the standard background risk statement with a requirement that labeling describe the background population rates of major birth defects and miscarriages regardless of drug exposure, as well as information—if available—for populations for which the drug is labeled. A section titled “Females and Males of Reproductive Potential” was also added.
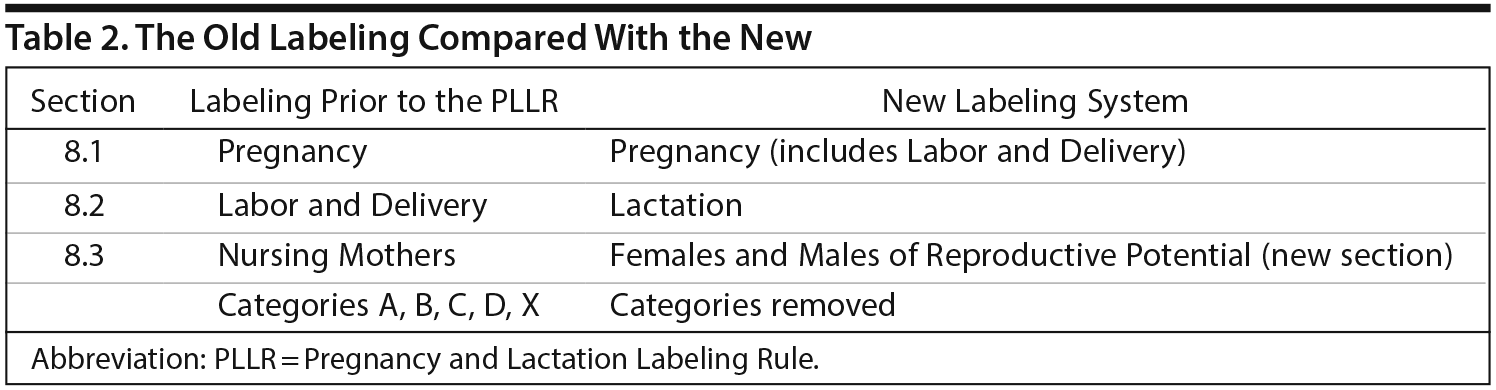
New PLLR Labeling vs the Old Labeling
Section 8 of drug labeling contains information regarding the use of a medication in specific populations. Dr Sahin compared the format of the new and old labeling and then explained the new format (Table 2). In the PLLR format, reproductive safety information is subdivided into 3 sections: Pregnancy, Lactation, and topics relating to Females and Males of Reproductive Potential. The Pregnancy and Lactation sections, in turn, are formatted to include 3 major subsections: Risk Summary, Clinical Considerations, and Data.
The new Pregnancy section (8.1) incorporates the old section on Labor and Delivery, previously section 8.2, and the new Lactation section (8.2) represents what was previously a section on Nursing Mothers (the old 8.3). In addition, a new section called Females and Males of Reproductive Potential (8.3) includes recommendations for pregnancy testing and/or contraception as well as any available data that suggest drug-associated fertility effects; Dr Sahin explained that previously, such information was dispersed throughout labeling and often hard to find.
Click figure to enlarge
The Pregnancy and Lactation sections have the same basic format, each with a Risk Summary, Clinical Considerations, and Data. In the Pregnancy section (8.1), pregnancy registry information, including contact information, is listed as the first item in order to encourage enrollment in pregnancy registries and improve data collection (Table 3). Dr Sahin reiterated that the Risk Summary portion of Section 8.1 describes the known risks of the product in the context of the background risk, both in the general population and in the disease population, of having a birth defect or adverse outcome regardless of drug exposure.

Click figure to enlarge
The Clinical Considerations portion of the Pregnancy section (8.1) covers disease-associated risk, maternal adverse reactions, and fetal/neonatal adverse reactions (eg, neonatal hypoglycemia, neonatal withdrawal syndrome). It also includes recommendations and summary information, if available, for dose adjustments and use of the product in labor and delivery. The Data portion provides the data that support the Risk Summary. Human data come first, taking precedence over animal data. Newly acquired human data will be added to labeling if it informs or changes the risk profile. Labeling will state when there are no human data available.
Advantages of the New Format
Drs Yao and Sahin explained that under the previous labeling system and its risk categories, which were too simplistic, prescribing decisions were often based on incorrect assumptions about safety in pregnancy. In the new labeling system, priority is placed on human data as well as timeliness, consideration of medical/disease factors, and background risk information. In the absence of background risk information about pregnancy outcomes in the general population and, if possible, in a group of women with a similar disease state but without the medication exposure, isolated data about exposure to a specific medication during pregnancy are difficult to interpret.
Major congenital malformations occur in 3%-4% of all pregnancies, for instance, and approximately 10% of all babies are born prematurely (Table 4). “Clinicians often don’ t appreciate when they read labeling that the background risk is never zero,” Dr Sahin said. “But there is always a risk of having a birth defect, miscarriage, or other adverse outcome. The background risk summary should help provide some context for a risk-benefit analysis,” as will information about disease-associated risk in the Clinical Considerations portion of the label. The intent overall is to provide prescribers with more relevant information for critical decision-making and to facilitate more comprehensive risk-benefit conversations with patients. Both officials explained that the PLLR provides a framework for more data collection in pregnant women and is a step toward more evidence-based prescribing. The rule brings greater focus, they said, to the importance of quantifying the reproductive safety of medications through more research.

Click figure to enlarge
Adoption of the PLLR
While the PLLR requires only those drugs approved since June 30, 2001, to move to the PLLR format, the FDA is encouraging manufacturers of older drugs to voluntarily convert to the new format, Dr Yao said. She estimated that about 10,000 products (including generic products) will need to have labeling converted (not including “voluntary” conversions) and said that the FDA’s review time will vary based on what information is available and on the due diligence that companies put into their labeling up front. The FDA is itself also working assiduously to review increasingly large numbers of applications.
When asked about responsibility for content in the labeling and for oversight of data accuracy, Dr Yao explained that drug labeling is owned by the sponsor and negotiated with the FDA. Ensuring as much consistency as possible across labels is the job of the FDA’s Pediatric and Maternal Health Division, the Labeling Development Team in the Office of New Drugs, and, in the case of psychoactive drugs, the Division of Psychiatry Products in the Office of New Drugs.
Dr Yao emphasized that every drug has unique issues that must be communicated clearly and that the FDA has not been interested in using standard statements or “fill-in-the-blank” template-driven language. Yet, crafting definitive and meaningful risk statements is not a simple task, especially when data are lacking and constantly evolving. Dr Yao pointed out that the 250-plus labeling changes that occurred in 2016 and just under 50 that occurred in 2015 made such challenges clear. When data are sufficient, she explained, the new format provides an excellent framework to discuss data and provide risk statements. But, when data are absent—or, even worse, when some data are available but the quality and quantity are limited—risk statements become difficult to compose in a way that can be easily interpreted by prescribers and patients.
Dr Yao shared that the agency created a PLLR working group to discuss the challenges of limited data; the group includes experts from across the FDA in clinical medicine, pharmacology-toxicology, clinical pharmacology, epidemiology, statistics, and legal counsel. In addition, a law signed in 2016 by President Barack Obama (21st Century Cures)7 called for the establishment of a federal task force to identify and address gaps in knowledge and research on safe and effective therapies for pregnant and lactating women and to report to Congress with specific recommendations. Dr Yao reported that the task force was being convened and that she is hopeful that a future report to Congress will advance the availability of high-quality pregnancy information.
In conclusion, Dr Yao emphasized that the PLLR is only the start. She described the PLLR as a house that currently is only sparsely furnished: “We’ ve got the framework now for a beautiful house. But now we must furnish the house. It is empty right now in many rooms, and we have to figure out how to furnish the house with the information we need.”
Insights From Pediatric Research/Labeling
Experiences in pediatric drug development and pediatric labeling may, in the meantime, offer a helpful model on how research that informs labeling for specific populations can be improved upon, Dr Yao said. Prior to 1997, over 80% of products contained no pediatric-specific information in labeling, and during the 1990-1997 period, fewer than a dozen clinical trials were ongoing for pediatric-specific therapeutics. Today, in contrast, fewer than half of the drugs available in the United States contain no pediatric-specific labeling or safety information. Dr Yao said she believes much of this progress can be credited to Congressional action and a combination of incentives and requirements.
The Best Pharmaceuticals for Children Act (BPCA),8 passed in 2002 (after 3 prior attempts since 1992), reauthorized in 2007, and permanently reauthorized in 2012, provides a financial incentive to companies to voluntarily conduct pediatric studies. The Pediatric Research Equity Act (PREA),9 passed in 2003, requires companies to assess the safety and effectiveness of certain products in pediatric patients. These 2 important statutes have worked well together to improve the level of pediatric-specific information, Dr Yao said.
BPCA and PREA advanced pediatric labeling beyond what the FDA had been able to do previously. Dr Yao pointed out that “The landscape changed considerably for pediatric-specific labeling because Congress got involved.” The legal decision was spurred by many advocacy groups and ultimately resulted in the passage of the BPCA and PREA and the permanent reauthorization of both laws in 2012.
The PLLR in the FDA’s Division of Psychiatry
Tiffany R. Farchione, MD, Deputy Director of the Division of Psychiatry Products, described her division’s approach to PLLR labeling changes, using sertraline hydrochloride (Zoloft) as an example of how older medications are converted under the new rule. When the PLLR went into effect in 2015, the division estimated that over 80 products would need to have labeling converted (only conversions, not including new drugs or supplements with efficacy data for new indications) and established a general timeline for these conversions for 2018-2020. A PLLR Advisory Group comprising a regulatory project manager, clinical reviewer, and nonclinical reviewer (who reviews animal data) was established within the division to develop a unified process for managing PLLR submissions. The group decided to aim for a 6-month total review period for each labeling conversion, including about 1 month for negotiations with the manufacturer.
Conversion of the Zoloft labeling illustrates how text from previous labeling must be overhauled, not just rearranged or reformatted. In the old labeling, Dr Farchione explained, the drug was given a Category C designation. Labeling included animal data and stated there were no adequate and well-controlled studies in humans; it did not address the first trimester. To convert the labeling, the FDA asked the manufacturer for a literature review of teratogenicity data (this had occurred prior to implementation of the final PLLR rule), and the FDA conducted its own concurrent review within the Division of Epidemiology. The reviews led to similar conclusions, and so the FDA and manufacturer entered into negotiations on labeling language.
The Risk Summary in the new labeling acknowledges the basic findings from published epidemiologic studies of pregnant women with first-trimester exposure and also offers a separate short summary of these studies with a discussion of some of the research limitations (eg, lack of control of confounders).
Studies/Methodologies to Inform Reproductive Safety
The PLLR establishes a new framework for describing available data and informing risk-benefit decision-making. Now, more data, and higher-quality data, are needed to populate the labeling framework. Pregnancy registries are considered the gold standard for studying the reproductive safety of medications and are highlighted in the new labeling. However, various types of studies, including prospective cohort studies and cross-sectional analyses of larger administrative databases, can provide important complementary information on pharmacotherapies during pregnancy.
Cohort Studies Using Secondary Data
Krista Huybrechts, MS, PhD, a pharmacoepidemiologist from Brigham and Women’s Hospital and Harvard Medical School, explained that cohort studies using secondary data can be a “productive and powerful” complement to pregnancy registries and other types of postmarketing research for evaluating drug safety during pregnancy. Typically, these studies link medical claims data, which include inpatient and outpatient diagnoses and procedures, and prescription claims data. Drug exposure information is documented before outcomes occur (ie, at the time the prescription is filled), and the recall bias that plagues case-control studies is not a concern. Cohort studies nested in administrative data often offer large sample sizes—and therefore the ability to study individual drugs rather than drug class—and allow the study and assessment of multiple exposures and outcomes. In addition, the use of secondary data is efficient in terms of cost and time.
However, several limitations and potential problems with the use of secondary data sources to assess drug safety during pregnancy, such as confounding, exposure misclassification, and outcome misclassification, must be considered and addressed through rigorous analyses, Dr Huybrechts explained. There may be limited information available on some potential confounders (eg, severity of illness, smoking, alcohol abuse, body mass index) and incomplete ascertainment of pregnancy terminations, for instance. In addition, prescription dispensing does not always equate to actual use of the medication or adherence to the prescribed regimen.
Dr Huybrechts discussed her work with the Medicaid Analytic eXtract (MAX) Pregnancy Cohort, a cohort of pregnancies resulting in live births nested in the national MAX database. To illustrate how the limitations of cohort studies using secondary data can be addressed, she referred to the statistical analyses undertaken for a MAX cohort study10 on first-trimester antipsychotic use in pregnancy and the risk for congenital malformations overall and cardiac malformations specifically.
Like other administrative databases, MAX is populated with claims data that cover demographics, diagnoses, procedures, and dispensed outpatient prescriptions. Since Medicaid covers close to 50% of births in the United States, the database offers a much larger dataset than pregnancy registries. Dr Huybrechts said that data for more than 1.5 million pregnancies have been linked to live-born infants for 2000-2013.
With any observational study, confounding is a significant concern—and indeed, “a lot of published studies [of medication exposure during pregnancy] fail to adjust for a broad range of potential confounding variables,” Dr Huybrechts said. In the cohort study on first-trimester antipsychotic use in pregnancy10 led by Huybrechts and colleagues, all point estimates were elevated in the crude, unadjusted analysis. Adjustment for psychiatric indications alone caused point estimates to shift toward the null, and a second level of adjustment accounting for about 50 potential confounding variables brought all associations to null (relative risk ≈ 1) except for 1 drug. Researchers then took it a step further to account for potential residual confounding by variables not well measured in administrative data. They utilized high-dimensional propensity score analyses to identify and include a large number of empirically identified covariates (in addition to the investigator-defined covariates) to serve as proxies for unmeasured variables. The size and richness of the data source made such an approach possible, she explained.
With prescription medication database studies, false-positives with regards to exposure to a drug are a concern, Dr Huybrechts said. A misclassification of a pregnancy to the “exposed” group can occur if a prescription was filled but not taken by the patient or if adherence was suboptimal. To evaluate for the potential impact of such exposure misclassification in the MAX Pregnancy Cohort, the investigators redefined exposure as having filled at least 2 prescriptions during the etiologically relevant window. They also strove for high specificity of the outcome definition. Because the ICD-9/10 diagnoses found in claims data are used to justify procedures and health services (not to serve research purposes), algorithms need to be carefully developed to define outcomes in a valid manner. With high specificity, the relative risk will be unbiased even when there is imperfect sensitivity as long as it is nondifferential, Dr Huybrechts explained. It is essential, she emphasized, to validate the outcome definition using medical record review.
While there is not a risk of control selection bias in cohort studies, there are risks of other selection biases. Because the MAX Pregnancy Cohort includes only live births, severe malformations that result in spontaneous abortions or stillbirths will be missed, as will pregnancy terminations for congenital anomalies. There is no perfect solution for reducing such selection biases, but it is possible and advisable to assess their impact using quantitative biases analyses with selection probabilities for exposed and unexposed pregnancies informed by the literature, Dr Huybrechts said.
At the end of her presentation, Dr Huybrechts described an initiative called the International Pregnancy Safety Study (InPreSS) Consortium, a collaboration among research groups with access to health care databases that have a demonstrated ability to study the safety of medications in pregnancy. The initiative combines large-scale pregnancy data from registries in Denmark, Finland, Iceland, Norway, and Sweden with pregnancy cohort data from the US Medicaid Analytic eXtract. A recent cohort study11 from the consortium looking at stimulants and the risk of congenital malformations included almost 3,000 exposed pregnancies, Dr Huybrechts said. Findings from the study, which covered more than 4.3 million pregnancies, were reported after the meeting, in December 2017.
Types of Human Data: Strengths and Weaknesses
Lockwood Taylor, PhD, Deputy Director of the FDA’s Division of Epidemiology II, explained that the FDA generally considers 3 sources of postmarketing human data for inclusion in labeling: case reports, pregnancy-exposure registries, and retrospective population-based studies. Each type of study design has its pros and cons.
Case reports. Case reports are either identified in the literature or submitted to the FDA Adverse Event Reporting System by drug manufacturers, patients, prescribers, and others. These may offer early signal detection—”red flags” for potential teratogens—and can be helpful for rare outcomes. However, they provide limited evidence for causal assessment and lack a denominator (the total number of pregnancies exposed from which cases arise). Case reports and case series are also confounded by comorbidities or coexposures, and their voluntary and retrospective reporting introduces biases.
Pregnancy registries. Pregnancy registries can be required by the FDA under certain circumstances. Registries can offer rigorous prospective assessment of outcomes during and after pregnancy. They can be helpful for hypothesis generation, and, if well-conducted and sufficiently powered, they may also rule out teratogenic effects, Dr Taylor noted. Among their disadvantages: traditionally low enrollment, potential selection bias due to the timing of enrollment in pregnancy and to the voluntary nature of enrollment, and a limited ability to adjust for confounding (especially when the sample size is small).
Retrospective population-based studies. Dr Taylor explained that population-based observational studies (retrospective) may also be required by the FDA to collect more information on risks of adverse events in pregnancies exposed to specific drugs. These include electronic claims-based studies/administrative databases, national registry studies, and, to a lesser extent, population-based case-control studies. A major advantage of this type of research is the potential for linking multiple data sources, Dr Taylor said. In addition, such studies may offer large datasets with appropriate comparison groups for detecting associations between specific drugs and outcomes. There are important disadvantages, however, and here Dr Taylor reiterated some of the challenges discussed by Dr Huybrechts. While studies involving electronic health care databases avoid both recall bias of drug exposure and self-selection bias, he said, there are issues with confounding and other challenges such as the determination of gestational age and the ascertainment of spontaneous and elective abortions.
The FDA’s Challenges in Interpreting Safety
The FDA faces a variety of challenges to interpreting safety of a drug overall and when used during pregnancy or lactation, Dr Taylor explained. The agency often must make labeling decisions based on observational data with considerable methodological limitations and variable quality. Moreover, findings often conflict, making it challenging to interpret and communicate risk for pregnancy labeling. The goal, he reiterated, is to inform physicians of potential risks to the patient and to provide clear and consistent information that assists providers in their risk/benefit decisions. “We need to be aware of [and guard against] the unintended consequences of labeling observational data,” such as false assumptions of causality and confusion over unclear or conflicting results, he said. “It’s also important to avoid situations in which a medical provider withdraws or switches their patient’s treatment based on biased study results and situations in which a pregnancy may be terminated based on undue alarm over biased study results.”
Dr Taylor used the new pregnancy labeling of sertraline to illustrate how the FDA arrived at a “bottom line” Risk Summary statement in the face of a variable and potentially confusing body of research. Approximately 10-15 epidemiologic studies had been published regarding selective serotonin reuptake inhibitor (SSRI) exposure during pregnancy, but the studies varied in quality and size. Several of these studies reported an observed increased risk of congenital cardiac defects, specifically septal defects, but there were concerns about study design and the validity of results. Fortunately, Dr Taylor said, “we came across a well-conducted meta-analysis that quantitatively summarized the results for us.” The FDA decided to include the main findings of this meta-analysis in the Data section of the pregnancy labeling for sertraline along with the summary odds ratios and confidence intervals. The labeling then briefly explains that most of the epidemiologic studies reporting an increased risk did not have comparison groups to allow for the control of important potential confounders. The FDA’s intent, Dr Taylor explained, was to include enough information—but not too much—and to acknowledge the problematic studies while putting them into perspective. “We needed to acknowledge the existence of the [lower-quality] studies but express skepticism about the validity of the results,” he said.
Ideally, the FDA would like to see more research on psychiatric drugs and specific defects, more sensitivity analyses, and the replication of results using a variety of study designs and data sources, Dr Taylor said. He also noted that the agency is open to suggestions from stakeholders on how best to craft Risk Summaries and shape the language when data are inconclusive.
MGH Pregnancy Registry for Atypical Antipsychotics
Lee S. Cohen, MD, Director of the Ammon-Pinizzotto Center for Women’s Mental Health at Massachusetts General Hospital, presented on the National Pregnancy Registry for Atypical Antipsychotics (NPRAA). Atypical antipsychotics, or second-generation antipsychotics, are being used by women of reproductive age across a growing number of indications, both FDA-approved and off-label. These illnesses and indications include schizophrenia, bipolar disorder, treatment-resistant depression, anxiety disorders, and insomnia. In contrast to older antipsychotics, which often caused elevated prolactin as an adverse event and decreased fertility, most second-generation antipsychotics are prolactin-sparing, meaning that fertility is much less likely to be impaired than it was with older typical antipsychotics. This is especially important given that an estimated 50% of pregnancies in the United States are unplanned.12
Incomplete data about the risks of fetal exposure to atypical antipsychotics prompted Cohen and his colleagues at Massachusetts General Hospital to establish the NPRAA13 about a decade ago, modeling it to a significant extent after the North American Antiepileptic Drug Pregnancy Registry.
“We really view our work as complementary to the work that’s done in other spaces,” such as large administrative claims-based data sources like the MAX Pregnancy Cohort,” Dr Cohen said. “We are approaching the problem from both angles.” The NPRAA and other pregnancy registries allow for prospective assessment of outcomes, rigorous determination of exposure to medications, and the verification of outcomes. They offer an opportunity to confirm signals appearing in administrative databases and to refine the findings from other data sources.
The primary aim of the MGH Registry is to prospectively evaluate rates of major malformations among infants exposed to atypical antipsychotics in utero. However, Dr Cohen emphasized, the registry is also able to look at secondary outcomes—obstetrical outcomes such as preterm labor, neonatal outcomes, and maternal health outcomes including weight gain across pregnancy and risk for gestational diabetes—with a level of detail that is not possible without access to medical records and other source documents.
In the registry, pregnant women between the ages of 18-45 years who have taken at least 1 atypical antipsychotic during pregnancy are enrolled. Notably, the registry recruits an internal comparison group of women with histories of psychiatric morbidity who may be taking other psychiatric medications but have not taken an atypical antipsychotic during pregnancy.
Prospective phone interviews are conducted at 3 time points during the subject’s study participation: As soon as possible after enrollment, at 7 months’ gestation, and 2-3 months postpartum. Around the time of the 7-month interview, permission is sought to request medical records from health care providers up to 6 months after delivery. This enables review of obstetric, labor and delivery, and newborn pediatric records. Dr Cohen explained that there is systematic review of potential congenital malformations and ultimate adjudication of suspected malformations by an independent and blinded dysmorphologist.
Prospective registries tend to have modest numbers of participants, and growth takes time, as does the gathering of outcomes data and procurement and review of medical records. Importantly, a scientific advisory board determines when data are to be released, based on the level of confidence regarding findings using statistical models along with the anticipated clinical impact of the findings. A report from the registry published in 201614 suggested that atypical antipsychotics as a class do not substantially raise the risk of major malformations. Confidence intervals were still wide, Dr Cohen noted, but he said that with increased numbers of participants, the confidence intervals are tightening substantially. The registry is expected to soon shift from reporting on aggregate data (findings across the class of medications) to releasing reports on specific medications, starting with quetiapine. Dr Cohen noted that about 90% of patients in the registry who are taking atypical antipsychotics have taken them consistently across pregnancy.
Regarding funding of the MGH registry, Dr Cohen explained that all manufacturers of psychotropic medications being assessed are approached to help fund a portion of the registry’s operating budget, an approach consistent with FDA guidance on establishing registries. In addition, each manufacturer’s sponsorship status and duration of support is posted on the MGH registry website (womensmentalhealth.org) for transparency. All medications in the studied classes are investigated regardless of support; however, the placement of registry information into product labeling does not imply financial support of that registry. Dr Cohen said that in his experience with the MGH registry, which has expanded to include antidepressants and stimulants, pharmaceutical companies appear to have varying perspectives on the importance of reproductive safety pharmacovigilance and supporting pregnancy exposure registries. The FDA strongly encourages companies to participate in pregnancy exposure registries but cannot necessarily require companies to participate in this endeavor. Dr Cohen said this appears to be interpreted differently by industry representatives, with some companies deciding to help fund their registry and others expressing a desire to opt out of financial support unless it is explicitly required.
In a later panel discussion featuring industry perspectives of pharmacovigilance and reproductive age women, several industry representatives shared their views of research, including that companies are in the process of learning more about the role of registries and that there is interest in methods for detecting teratogenic signals as early as possible. Global efforts such as the InPreSS international research consortium (described by Dr Huybrechts) were praised as important for increasing statistical power, and support was expressed for a multifaceted research that includes both exposure registries, which are labor-intensive but can provide high-quality information, and secondary analysis of epidemiologic data, which can offer a fast track for the detection of early signals for teratogens.
The Maternal Side of the Equation
The risk-benefit analysis concerning the use or discontinuation of psychiatric medications during pregnancy is not only about the fetus, but also about maternal mental health, and, far too often, people forget to account for the risks of untreated psychiatric illness. To illustrate the importance of maternal mental health considerations, Dr Cohen pointed to 2 studies of disease recurrence risk with continued or discontinued treatment during pregnancy.
The first,15 which looked at relapse of bipolar disorder in patients who were euthymic at conception, showed that recurrence risk was 2.3 times greater after discontinuation of mood stabilizer
treatment than with continued treatment.
The second study,5 which focused on patients with unipolar major depression, found that those who discontinued medication relapsed 5 times more frequently over the course of their pregnancies compared with those who maintained their medication.
What is in labeling and what is communicated to patients have significant implications for maternal mental health, Dr Cohen emphasized. Balanced consideration of both the relative risks of fetal exposure to medications and the risks of recurrence with treatment discontinuation is essential, he said.
Pregnancy Registries: The FDA’s “Gold Standard”
The FDA Amendments Act of 2007 gave the agency authority to require postapproval studies to assess serious risks such as birth defects, and pregnancy registries “are the most common type” of postmarketing requirement/postmarketing commitment, according to Dr Sahin of the FDA. Pregnancy registries are considered the gold standard in collection of postmarketing safety data during pregnancy, she said, because they offer an efficient way to begin data collection immediately after drug approval, provide detailed patient-level data on medication exposure and covariates, and can allow validation of outcomes based on medical record review and expert assessment.
Dr Sahin emphasized that data collection is a shared responsibility among all stakeholders. In 2014, in preparation for an FDA public meeting about postmarketing safety studies in pregnant women, the FDA conducted a review of 59 products from 38 pregnancy registries listed on the FDA Pregnancy Registry Webpage. Half of the registries were required by FDA and half were voluntary.
Stakeholders from different perspectives who came to the meeting emphasized the need for data and had several key messages. One was that a combination of approaches can overcome the limitations of individual study designs and increase confidence in consistent findings. Another was that multiproduct or disease-based registries have generally been more successful for collecting data and sustaining registries than registries of a single drug; this is because an existing infrastructure can be leveraged to pool resources and accommodate newly approved drugs. In addition, processes are streamlined for providers and patients, and a greater number of participants are enrolled, increasing the opportunity to yield clinically meaningful data.
A good example of a multidrug/disease pregnancy registry is the Antiretroviral Pregnancy Registry (http://apregistry.com), Dr Sahin said. In 2016, the registry was collecting data on 53 drugs, was supported by 27 manufacturers, and had collected data on over 17,000 exposed live births (including more than 8,000 first-trimester exposures). Seventy countries were participating, with over three-quarters of the reports coming from the United States.
Disseminating Information on the PLLR
Patients as the “End Users”
Most women—84% in one internet-based survey16—use multiple information sources when seeking information about medicine use during pregnancy, and in one-quarter of these cases, women find conflicting information, which often leads to anxiety and to decisions to discontinue a medication during pregnancy. Ruta Nonacs, MD, PhD, Editor-in-Chief of www.womensmentalhealth.org based at MGH, said that, in discussing how reproductive safety and information in the PLLR can best be disseminated, it is important to consider not only how physicians obtain medical information and make decisions, but also how patients, who are the end users, do as well.
A growing number of women of reproductive age have attained better health and a higher quality of life with use of medications than was possible in the past and are able to pursue pregnancies. Dr Nonacs referred to a study,17 also cited by Dr Yao, showing that first trimester use of prescription medication increased by more than 60% between 1976 and 2008 and that pregnant women take an average of 2.6 medications at any time during pregnancy.
Motivated patients seek opinions from multiple providers who often offer conflicting advice, Dr Nonacs said. For example, a newly pregnant patient who has a history of depression and anxiety and has been stable on antidepressant medications for many years will likely seek the option of her psychiatrist, her obstetrician, and her primary care provider. In what is a common scenario, Dr Nonacs explained, each physician will offer different advice and conflicting information, and the patient will be left feeling alone and confused. When available, reproductive psychiatrists, in advising patients regarding the reproductive safety of psychiatric medications, often play the role of an arbiter in this situation.
Patients also use internet resources, consult with family and friends (who may not understand the risks of the untreated illness or the baseline risk of major malformations in the general population), and face emotional factors and pressures to do everything “right” during their pregnancies. Some patients make assumptions that since their doctor prescribed the medication, it is safe to use during pregnancy. Dr Nonacs emphasized that the internet is often an important source of information, but it can be misleading. Worrisome findings tend to gain more coverage in the media and rise to the top of search results, and out-of-date but popular articles can persist in cyberspace. This is of concern as the literature in this area continues to evolve.
A study18 of YouTube videos as a source of information on medication use in pregnancy found that 67% were from law firms and that SSRIs were the most common class of medication named (88% of videos on SSRIs indicated that they were unsafe in pregnancy). Some internet sites offer “safe lists” that are misleading. One study19 of such “safe lists” showed that for 40% of the medications listed there were no or very limited data on reproductive safety; moreover, medications not on these lists are not necessarily “unsafe,” Dr Nonacs said.
A Patient Perspective
A patient who experienced postpartum depression and anxiety after the birth of her first child spoke at the meeting about her experience of a delay in getting a diagnosis (after seeking help during numerous postpartum visits to the hospital), her wait time for an appointment with a perinatal psychiatrist, the difficulty she faced in finding helpful safety information online regarding medication and breastfeeding, and the difficulty in navigating the decision about whether to use a medication during her next pregnancy and while breastfeeding. She eventually found a team of providers who worked together and whom she could trust, she explained. But she noted that the process was so difficult and stressful that she decided to start an advocacy group and peer-to-peer support network in her local area for women with postpartum depression and/or anxiety. Drug labels, especially their (old) letter categories, can be terrifying to patients who are anxious to begin with about the health of their child, and labeling language can mistakenly drive some health care providers to limit treatment choices, she told meeting participants. She also shared a story about a friend who committed suicide after being unsuccessful in finding treatment for postpartum depression and being told that breastfeeding narrowed her medication treatment options to 1 drug that did not help her. Having an educated clinical care team and peer-to-peer support are critical, she said.
Clinicians’ Sources of Information
Where do physicians get their information? Research has shown that for information about patient diagnosis and treatment, professional journals rank most highly, along with continuing medical education (CME) programs and communication with colleagues. Dr Nonacs said that a sizable number of physicians also conduct regular Google searches to find medical information. Among physicians’ resources for drug reproductive safety information in addition to package inserts are free sites such as WebMD and Drugs.com, paid sites such as UpToDate and Epocrates, and the electronic medical record. Just like popular patient sites and blogs, even medical sites and CME activity pages often feature negative headlines that are quickly amplified on the internet, Dr Nonacs noted.
COMMUNICATION STRATEGIES AND OPPORTUNITIES FOR COLLABORATION
Dr Nonacs urged that more emphasis must be placed in medical practice on planning in advance for pregnancy. Approximately 50% of pregnancies are unplanned, and obstetricians typically first see patients during a pregnancy at 8-10 weeks after conception. Just as the US Preventive Services Task Force now recommends that all women of reproductive age take supplemental folic acid to decrease the risk of birth defects, all women with chronic or recurrent illness who are taking medication should discuss contraception and review plans for potential pregnancy—including the need to communicate early on, before conception occurs, about the use of medications during pregnancy. “When we pull out the prescription pad, we should be thinking, Does this woman have contraception?” Dr Nonacs said. “We should tell her, ‘ If you’ re thinking about getting pregnant, let’s talk about it early.’ ” The weighing of risks and benefits is a time-intensive and individual process that includes questions such as Does this patient need to stay on the medication? What are the risks of untreated illness? What are the risks of the medication during pregnancy? and Are there safer alternatives?
As with other medical information, PLLR information should be clearly communicated, easy to understand, easy to access, and consistent. Dr Nonacs said she believes that centralization of medical information is generally helpful for clinicians, who currently can get pieces of information from varying places without always clearly knowing the sources, timing, or scientific references of that information. Information about the safety of medications in pregnancy is no exception. In that light, Dr Nonacs explained, transparency about scientific references is an important principle as stakeholders work to implement and incorporate the PLLR into clinical care. The electronic medical record is currently onerous for many clinicians, she noted, but it holds potential for drawing together resources and information to help guide decision-making.
Dr Nonacs said that increasing awareness and understanding of the PLLR will involve both traditional and innovative communication strategies. “Clinicians have had a language—of letter categories—that we gave them for talking about safety, and now we’ ve taken it away from them,” she said. The FDA has a variety of communication tools that have been and can be harnessed for disseminating information on reproductive safety and the PLLR, from webinars, podcasts, and other educational outreach activities to consumer updates to e-mail Listservs and social media.
Opportunities for the FDA to collaborate include working with professional organizations such as the American College of Obstetricians and Gynecologists and the Organization of Teratology Information Specialists, and with government agency activities/campaigns such as the Centers for Disease Control and Prevention’s Treating for Two, the National Institute of Health’s TOXNET, and WomensHealth.gov (Department of Health and Human Services). The nonprofit organization MotherToBaby produces helpful fact sheets about exposures during pregnancy and breastfeeding, Nonacs noted. Information dissemination is vital, meeting participants emphasized during a discussion period. “Clinicians get scared of pregnancy information,” one participant said. “We need to empower them to present this information . . . to be prepared and confident to use this information and translate it to their patients.”
Representatives from industry and other meeting participants emphasized that obstetricians and primary care physicians are often on the frontlines of reproductive safety discussions and decision-making and that patients need to have confidence in their physicians’ knowledge and guidance. One meeting participant shared her belief in the value of face-to-face CME meetings (compared with online courses), which often benefit from industry support, and others spoke of a need for more education about reproductive safety in residency programs. The need for academia and industry to find further ways to work together on education and information dissemination was also expressed.
One industry leader shared her belief that all parties—patients, physicians, and industry—need education about the new labeling system. In her company, she said, in-house medical affairs staff have fielded questions from patients and providers who are frustrated and who want more information about what is in labeling, what data are available, and what the labeling means for their own situation. In response to a question about measuring the impact of the PLLR, Dr Yao of the FDA said that it will be easy to measure rates and timing of labeling conversions but challenging to measure and assess the actual implementation of the PLLR in practice and its impact on patient-provider communication and decision-making. She reiterated that the sounder the data, and the better their quality and quantity, the easier it will be to communicate safety information to patients.
Published online: July 17, 2018.
REFERENCES
1. Food and Drug Administration, HHS. Content and format of labeling for human prescription drug and biological products; requirements for pregnancy and lactation labeling: final rule. Fed Regist. 2014;79(233):72063-72103. PubMed
2. Bateman BT, Huybrechts KF, Fischer MA, et al. Chronic hypertension in pregnancy and the risk of congenital malformations: a cohort study. Am J Obstet Gynecol. 2015;212(3):337.e1-337.e14. PubMed CrossRef
3. Hamilton BE, Hoyert DL, Martin JA, et al. Annual summary of vital statistics: 2010-2011. Pediatrics. 2013;131(3):548-558. PubMed CrossRef
4. Bardenheier BH, Imperatore G, Gilboa SM, et al. Trends in gestational diabetes among hospital deliveries in 19 US States, 2000-2010. Am J Prev Med. 2015;49(1):12-19. PubMed CrossRef
5. Tosato S, Albert U, Tomassi S, et al. A systematized review of atypical antipsychotics in pregnant women: balancing between risks of untreated illness and risks of drug-related adverse effects. J Clin Psychiatry. 2017;78(5):e477-e489. PubMed CrossRef
6. Santangeli L, Sattar N, Huda SS. Impact of maternal obesity on perinatal and childhood outcomes. Best Pract Res Clin Obstet Gynaecol. 2015;29(3):438-448. PubMed CrossRef
7. 21st Century Cures Act, 42 USC §2041 (2015).
8. Best Pharmaceuticals for Children Act, 42 USC (2002).
9. Pediatric Research Equity Act of 2003, 21 USC (2003).
10. Huybrechts KF, Hernández-D×az S, Patorno E, et al. Antipsychotic use in pregnancy and the risk for congenital malformations. JAMA Psychiatry. 2016;73(9):938-946. PubMed CrossRef
11. Huybrechts KF, Bröms G, Christensen LB, et al. Association between methylphenidate and amphetamine use in pregnancy and risk of congenital malformations: a cohort study from the International Pregnancy Safety Study Consortium. JAMA Psychiatry. 2018;75(2):167-175. PubMed CrossRef
12. Centers for Disease Control and Prevention. Unintended pregnancy prevention. CDC website. https://www.cdc.gov/reproductivehealth/unintendedpregnancy/index.htm. Accessed June 19, 2018.
13. Cohen LS, Viguera AC, McInerney KA, et al. Establishment of the National Pregnancy Registry for Atypical Antipsychotics. J Clin Psychiatry. 2015;76(7):986-989. PubMed CrossRef
14. Cohen LS, Viguera AC, McInerney KA, et al. Reproductive safety of second-generation antipsychotics: current data from the Massachusetts General Hospital National Pregnancy Registry for Atypical Antipsychotics. Am J Psychiatry. 2016;173(3):263-270. PubMed CrossRef
15. Viguera AC, Whitfield T, Baldessarini RJ, et al. Risk of recurrence in women with bipolar disorder during pregnancy: prospective study of mood stabilizer discontinuation. Am J Psychiatry. 2007;164(12):1817-1824, quiz 1923. PubMed CrossRef
16. Hפmeen-Anttila K, Nordeng H, Kokki E, et al. Multiple information sources and consequences of conflicting information about medicine use during pregnancy: a multinational Internet-based survey. J Med Internet Res. 2014;16(2):e60. PubMed CrossRef
17. Mitchell AA, Gilboa SM, Werler MM, et al; National Birth Defects Prevention Study. Medication use during pregnancy, with particular focus on prescription drugs: 1976-2008. Am J Obstet Gynecol. 2011;205(1):51.e1-51.e8. PubMed CrossRef
18. Hansen C, Interrante JD, Ailes EC, et al. Assessment of YouTube videos as a source of information on medication use in pregnancy. Pharmacoepidemiol Drug Saf. 2016;25(1):35-44. PubMed CrossRef
19. Peters SL, Lind JN, Humphrey JR, et al. Safe lists for medications in pregnancy: inadequate evidence base and inconsistent guidance from Web-based information, 2011. Pharmacoepidemiol Drug Saf. 2013;22(3):324-328. PubMed CrossRef
![]() Save
Save
![]() Share
Share
![]() Cite
Cite
Advertisement
GAM ID: sidebar-top