Objective: To evaluate relapse rates and predictors of relapse in 2 randomized, placebo-controlled trials of desvenlafaxine for major depressive disorder (MDD).
Method: Study 1: week 8 responders to open-label desvenlafaxine 50 mg/d entered a 12-week open-label stability phase. Patients with a continuing, stable response at week 20 were randomly assigned to 6-month, double-blind treatment (desvenlafaxine 50 mg/d or placebo). Study 1 was conducted between June 2009 and March 2011 at 87 sites worldwide. Study 2: week 12 responders to open-label desvenlafaxine 200 or 400 mg/d were randomly assigned to 6-month, double-blind treatment (desvenlafaxine 200 mg/d, 400 mg/d, or placebo). Study 2 was conducted between June 2003 and August 2005 at 49 sites in Europe, the United States, and Taiwan. Relapse was assessed separately by study with log-rank test using protocol definitions of relapse and with 17-item Hamilton Depression Rating Scale (HDRS-17) score ≥ 16 at any time during the double-blind phase. Kaplan-Meier estimates evaluated time to relapse, censoring data at months 1, 2, and 3 and overall; treatments were compared using hazard ratios. Cox proportional hazards models assessed relapse predictors.
Results: Overall relapse rates for all definitions were significantly lower for desvenlafaxine versus placebo for both studies (all P ≤ .002). In study 1, rates were significantly lower for desvenlafaxine versus placebo at month 2 (P = .016) and month 3 (P = .007) using the protocol definition. In study 2, relapse rates were significantly lower for desvenlafaxine versus placebo at months 1, 2, and 3 for both definitions (P < .0001-.002). Hazard ratios were similar at months 1, 2, and 3 and overall for both studies (0.382-0.639).
Conclusions: Desvenlafaxine 50 to 400 mg/d effectively prevented relapse at 6 months. Desvenlafaxine significantly prevented relapse early (month 1) versus placebo only in study 2.
Trial Registration: ClinicalTrials.gov identifiers: NCT00887224 and NCT00075257
An Analysis of Relapse Rates and Predictors of Relapse
in 2 Randomized, Placebo-Controlled Trials of Desvenlafaxine
for Major Depressive Disorder
ABSTRACT
Objective: To evaluate relapse rates and predictors of relapse in 2 randomized, placebo-controlled trials of desvenlafaxine for major depressive disorder (MDD).
Method: Study 1: week 8 responders to open-label desvenlafaxine 50 mg/d entered a 12-week open-label stability phase. Patients with a continuing, stable response at week 20 were randomly assigned to 6-month, double-blind treatment (desvenlafaxine 50 mg/d or placebo). Study 1 was conducted between June 2009 and March 2011 at 87 sites worldwide. Study 2: week 12 responders to open-label desvenlafaxine 200 or 400 mg/d were randomly assigned to 6-month, double-blind treatment (desvenlafaxine 200 mg/d, 400 mg/d, or placebo). Study 2 was conducted between June 2003 and August 2005 at 49 sites in Europe, the United States, and Taiwan. Relapse was assessed separately by study with log-rank test using protocol definitions of relapse and with 17-item Hamilton Depression Rating Scale (HDRS-17) score ≥ 16 at any time during the double-blind phase. Kaplan-Meier estimates evaluated time to relapse, censoring data at months 1, 2, and 3 and overall; treatments were compared using hazard ratios. Cox proportional hazards models assessed relapse predictors.
Results: Overall relapse rates for all definitions were significantly lower for desvenlafaxine versus placebo for both studies (all P ≤ .002). In study 1, rates were significantly lower for desvenlafaxine versus placebo at month 2 (P = .016) and month 3 (P = .007) using the protocol definition. In study 2, relapse rates were significantly lower for desvenlafaxine versus placebo at months 1, 2, and 3 for both definitions (P < .0001–.002). Hazard ratios were similar at months 1, 2, and 3 and overall for both studies (0.382–0.639).
Conclusions: Desvenlafaxine 50 to 400 mg/d effectively prevented relapse at 6 months. Desvenlafaxine significantly prevented relapse early (month 1) versus placebo only in study 2.
Trial Registration: ClinicalTrials.gov identifiers: NCT00887224 and NCT00075257
Prim Care Companion CNS Disord 2015;17(1):doi:10.4088/PCC.14m01681
© Copyright 2015 Physicians Postgraduate Press, Inc.
Submitted: May 20, 2014; accepted October 23, 2014.
Published online: February 26, 2015.
Corresponding authors: Roger S. McIntyre, MD, FRCPC, Department of Psychiatry and Pharmacology, University of Toronto, Mood Disorders Psychopharmacology Unit, University Health Network, 399 Bathurst St,
MP 9-325, Toronto, ON M5T 2S8, Canada ([email protected]),
and Matthieu Boucher, PhD, Medical Affairs, Pfizer Canada, Inc,
17300 Transcanada Highway, Kirkland, QC, H9J 2M5, Canada
([email protected]).
Incomplete resolution of depressive symptoms with treatment of major depressive disorder (MDD) is associated with early relapse and recurrence of depressive episodes.1,2 Risk of relapse is significantly reduced by continuation of antidepressant therapy after response to acute treatment.3 Treatment guidelines recommend that patients continue antidepressant treatment for at least 4 to 9 months after successful acute phase therapy to prevent relapse of a depressive episode.4–9 Antidepressant prescribing records in the United Kingdom from the 1990s and 2000s indicate that there is a trend toward longer durations of antidepressant treatment in recent years,10,11 but treatment duration remains below the guideline recommendations for many patients. In recent retrospective database analyses, 54% to 56% of patients with new antidepressant prescriptions had a treatment duration of at least 3 months,12,13 and antidepressant treatment for at least 6 months was reported for just 32.5% to 44.3% of patients.12–14
The recommended range for treatment duration for a depressive episode is substantial, and clinical decisions regarding how long to continue an individual patient on an antidepressant medication after acute phase resolution of symptoms must take into account the history and severity of that patient’s illness.4–9 The effectiveness of antidepressants in studies assessing maintenance of acute benefit with different durations of treatment could also instruct clinical recommendations. The serotonin-norepinephrine reuptake inhibitor desvenlafaxine (administered as desvenlafaxine succinate)15,16 has been demonstrated to be effective for the prevention of the relapse of depression at doses of 50 mg/d and 200 mg/d to 400 mg/d in separate double-blind, placebo-controlled studies of different designs.17,18 The objective of the current analysis was to evaluate rates and timing of relapse in 2 randomized, placebo-controlled trials of desvenlafaxine for the treatment of MDD. Predictors of relapse were also examined.
METHOD
Two previously published17,18 phase 3, multicenter, double-
blind, placebo-controlled, randomized, withdrawal, parallel-group studies were compared in this analysis (ClinicalTrials.gov identifiers: NCT00887224 and NCT00075257). Study 117 was conducted between June 2009 and March 2011 at 87 sites worldwide, including sites in North America, South America, Europe, and South Africa. Study 218 was conducted between June 2003 and August 2005 at 49 sites in Europe, the United States, and Taiwan. Study sites included academic and nonacademic sites, psychiatric practices, and research centers. The study protocols and any amendments received institutional review board or independent ethics committee approval, and both studies were conducted in accordance with the International Conference on Harmonization Guideline for Good Clinical Practice and the ethical principles that have their origin in the Declaration of Helsinki. Written informed consent was obtained from all participants before any protocol-required procedures were performed.
Study Design
Each study consisted of a screening period, an open-label treatment period, and a 6-month, double-blind, placebo-controlled, randomized withdrawal period (Figure 1). The duration of the open-label treatment period was 20 weeks in study 1 and 12 weeks in study 2. In study 1, the open-label period consisted of 2 phases: response and stability. All patients enrolled in study 1 received 8 weeks of open-label treatment with desvenlafaxine 50 mg/d (response phase). Patients who achieved treatment response (defined per study 1 protocol as 17-item Hamilton Depression Rating Scale19 [HDRS-17] total score ≤ 11 and Clinical Global Impressions Scale–Improvement20 [CGI-I] score ≤ 2) at week 8 received open-label desvenlafaxine 50 mg/d for an additional 12 weeks (stability phase). Patients with a continued stable response at the end of week 20 (HDRS-17 total score ≤ 11 and CGI-I score ≤ 2 and no HDRS-17 total score ≥ 16 or CGI-I score ≥ 4 at any visit during the stability phase) were randomly assigned 1:1 to receive 6 months of double-blind treatment with placebo or desvenlafaxine 50 mg/d.
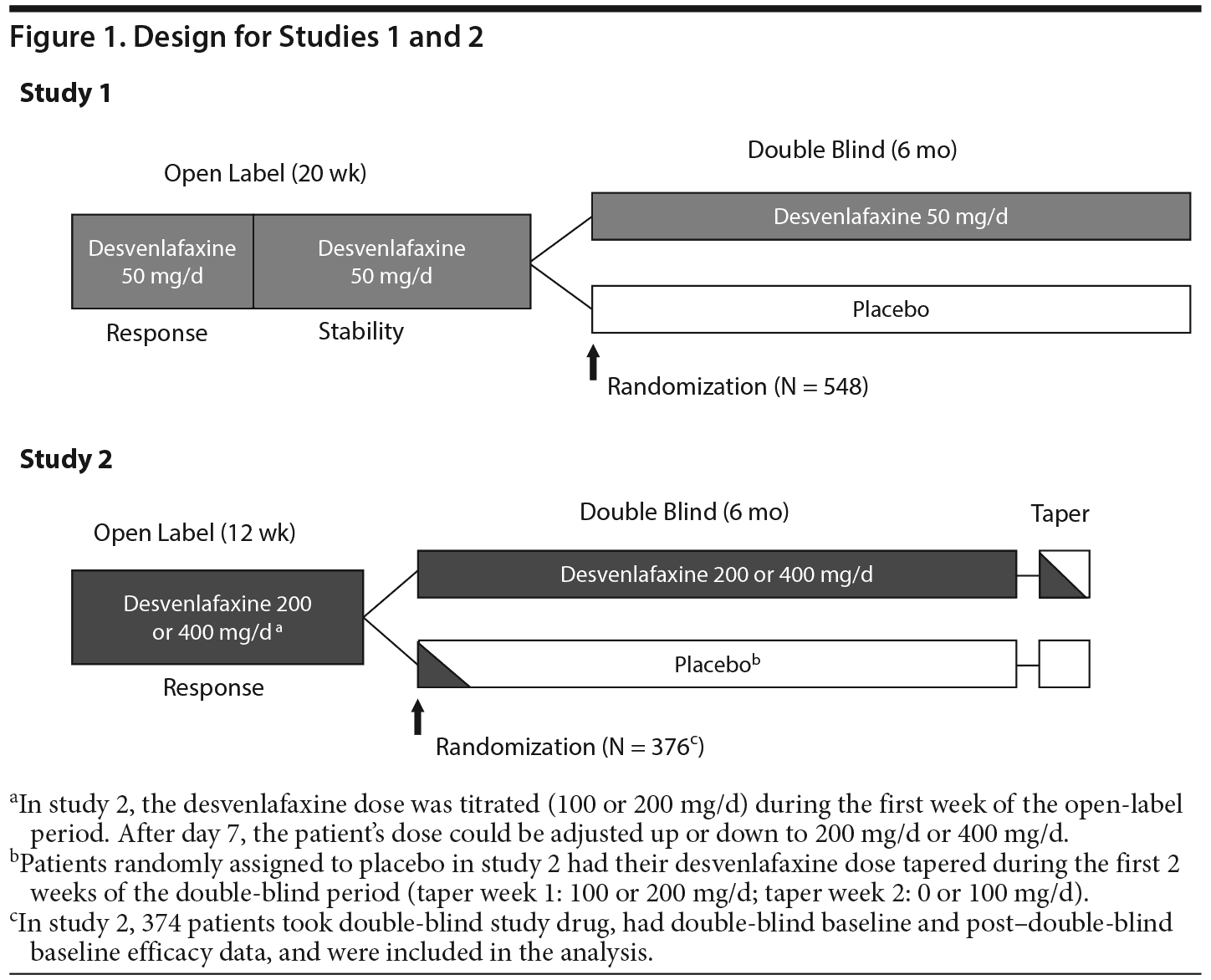
Study 2 included only a response phase in the open-label period. All enrolled patients received 12 weeks of open-label treatment with desvenlafaxine 200 or 400 mg/d (based on efficacy and tolerability at the investigator’s discretion). Responders (defined per study 2 protocol as HDRS-17 total score ≤ 11) at week 12 were randomly assigned to 6 months of double-blind treatment with desvenlafaxine or placebo. Patients assigned to desvenlafaxine received 200 or 400 mg/d based on their desvenlafaxine dose at the end of the open-label period. Patients assigned to placebo received desvenlafaxine 200 mg/d during week 1 of the double-blind period, 100 mg/d during week 2, and placebo starting at week 3.
For this post hoc analysis, relapse was assessed using the studies’ per-protocol definitions and—because those differed between the studies—a common definition based on HDRS-17 total score only. The per-protocol definitions were as follows: for study 1 only, HDRS-17 total score ≥ 16 at any time during the double-blind phase, study discontinuation due to unsatisfactory response, or hospitalization for depression, suicide, or suicide attempt; for study 2 only, HDRS-17 total score ≥ 16 at any visit, CGI-I score (versus double-blind baseline) ≥ 6, or study discontinuation due to unsatisfactory response. The HDRS-17 definition of relapse used for both studies was a HDRS-17 total score ≥ 16 at any time during the double-blind phase.
Patients
The patient populations for each study are described elsewhere.17,18 Briefly, both study 1 and study 2 enrolled adult outpatients (aged ≥ 18 years; ≤ 75 years for study 2) with a primary diagnosis of MDD based on criteria consistent with the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision21 and depressive symptoms for at least 30 days before screening. All enrolled patients had a HDRS-17 total score ≥ 20, a HDRS-17 item 1 (depressed mood) score ≥ 2, and a CGI–Severity of Illness20 (CGI-S) score ≥ 4 at screening and baseline.
Major exclusion criteria for each study included treatment with desvenlafaxine at any time in the past, known or suspected sensitivity to venlafaxine, and significant risk of suicide (based on clinical judgment or a HDRS-17 score > 3 on item 3 [suicide] at screening). Patients were excluded if they had current (within 12 months from baseline) manic episodes, posttraumatic stress disorder, obsessive-compulsive disorder, or clinically important personality disorder as assessed by the modified Mini-International Neuropsychiatric Interview22; depression associated with an organic mental disorder due to a general medical condition or neurologic disorder; history of a seizure disorder; or clinically important medical disease.
Statistical Analysis
Because studies 1 and 2 used different study designs and doses of desvenlafaxine, they were not pooled; instead, a side-by-side analysis was conducted. All analyses were performed for protocol and HDRS-17 definitions of relapse. The analysis populations were all patients randomly assigned to double-blind treatment (all randomized) for study 1, and all randomly assigned patients who took at least 1 dose of double-blind study drug and had a double-blind baseline and at least 1 post–double-blind baseline primary efficacy evaluation (intent to treat) for study 2.
Relapse was assessed using log-rank test for each study separately. Kaplan-Meier estimates evaluated time to relapse, censoring data at months 1, 2, and 3 and overall (month 6). Hazard ratios compared treatments overall and for data censored at months 1, 2, and 3. Predictors of relapse were assessed using Cox proportional hazards models for 3 sets of predictors. Model 1 included treatment and baseline characteristics and disease severity (age, gender, duration of current episode, number of prior MDD episodes [collected in study 1 only], and HDRS-17 total score at double-blind baseline), model 2 included baseline treatment and body mass index (BMI) category (normal [BMI ≤ 25 kg/m2], overweight [25 kg/m2 < BMI ≤ 30 kg/m2], or obese [BMI > 30 kg/m2]), and model 3 included treatment and number of nonstudy medications taken prior to or on day 1 of the double-blind period.
RESULTS
Patients
In study 1,17 874 patients entered the 8-week open-label response phase; 752 completed the response phase and 659/874 (75.4%) entered the open-label stability phase. A total of 576 patients completed the stability phase; 548/874 (62.7%) had a stable response to treatment, were randomly assigned to desvenlafaxine 50 mg/d (n = 272) or placebo (n = 276), and were included in the current analysis. In study 2,18 603 patients entered the 12-week open-label period, and 411 completed open-label treatment. In all, 376/603 patients (62.4%) were responders at week 12 and were randomly assigned to double-blind treatment; 374 (62.0%) took at least 1 dose of double-blind study drug, had double-blind baseline and post–double-blind baseline efficacy data, and were included in the current analysis (desvenlafaxine 200 or 400 mg/d: n = 189; placebo: n = 185).
Demographic and double-blind baseline characteristics for the 2 studies are listed in Table 1. Patients were 70% female and ranged in age from 18 to 77 years; 87% were white. The mean HDRS-17 total score at randomization was 4.6 for study 1 and 5.5 for study 2. No significant differences between treatment groups were observed for either study.
Relapse Rates
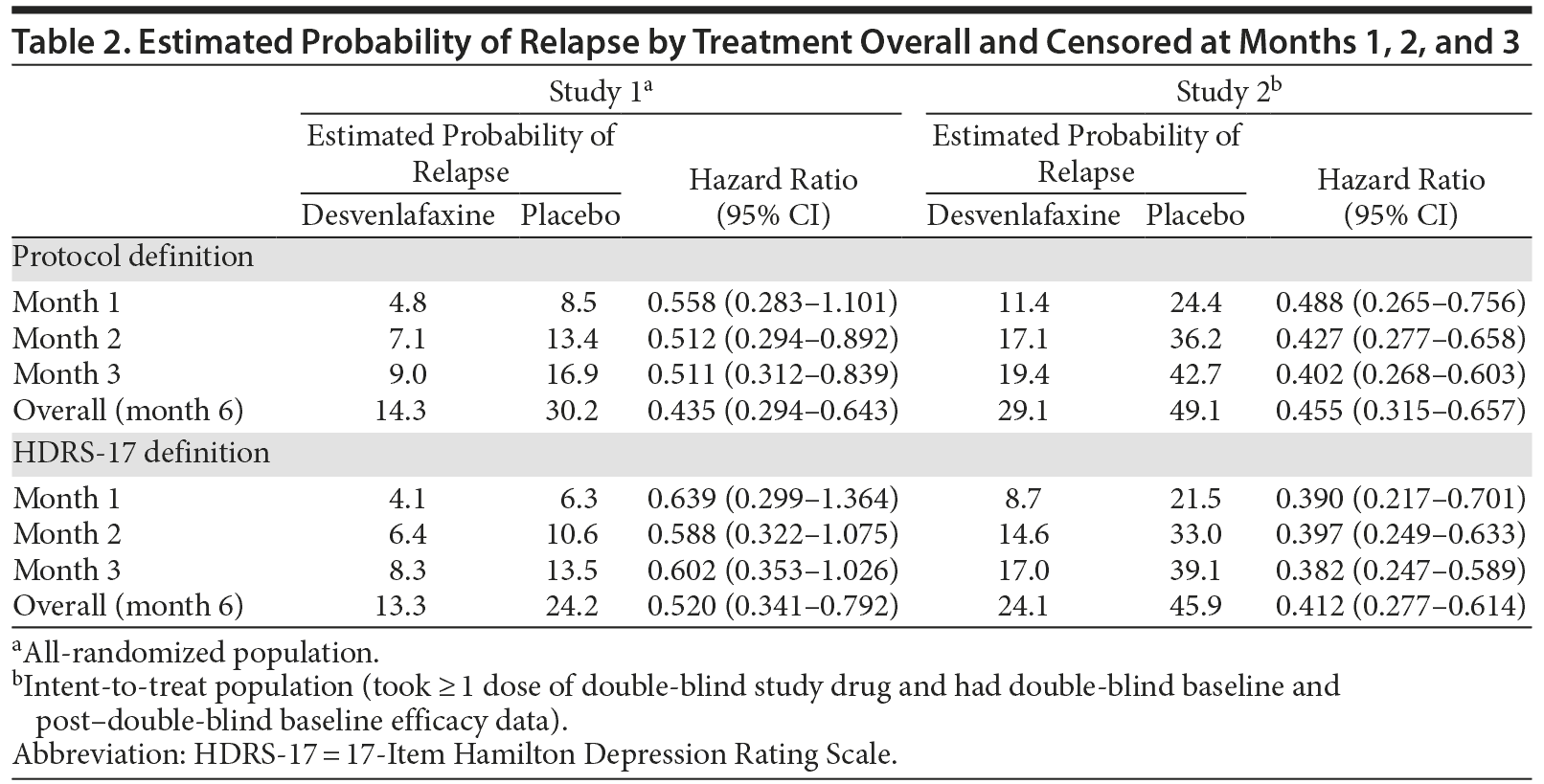
The estimated probability of relapse over the entire double-blind period based on either protocol or HDRS-17 definitions was significantly lower for desvenlafaxine compared with placebo in studies 1 and 2 (all P ≤ .0019). Rates of relapse were numerically lower in study 1 than in study 2 for both the desvenlafaxine and placebo groups, regardless of the definition of relapse (Figure 2 and 3).
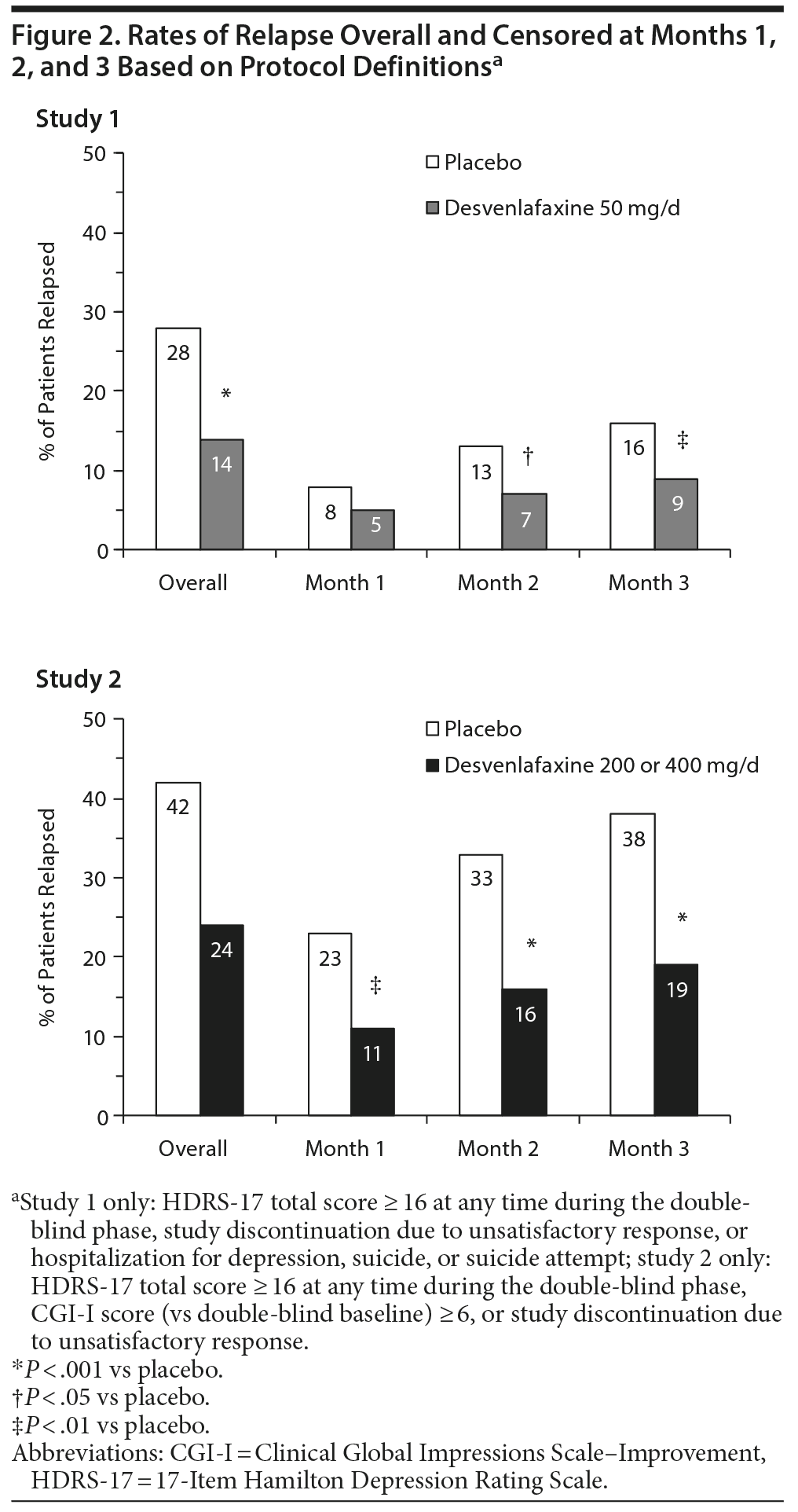
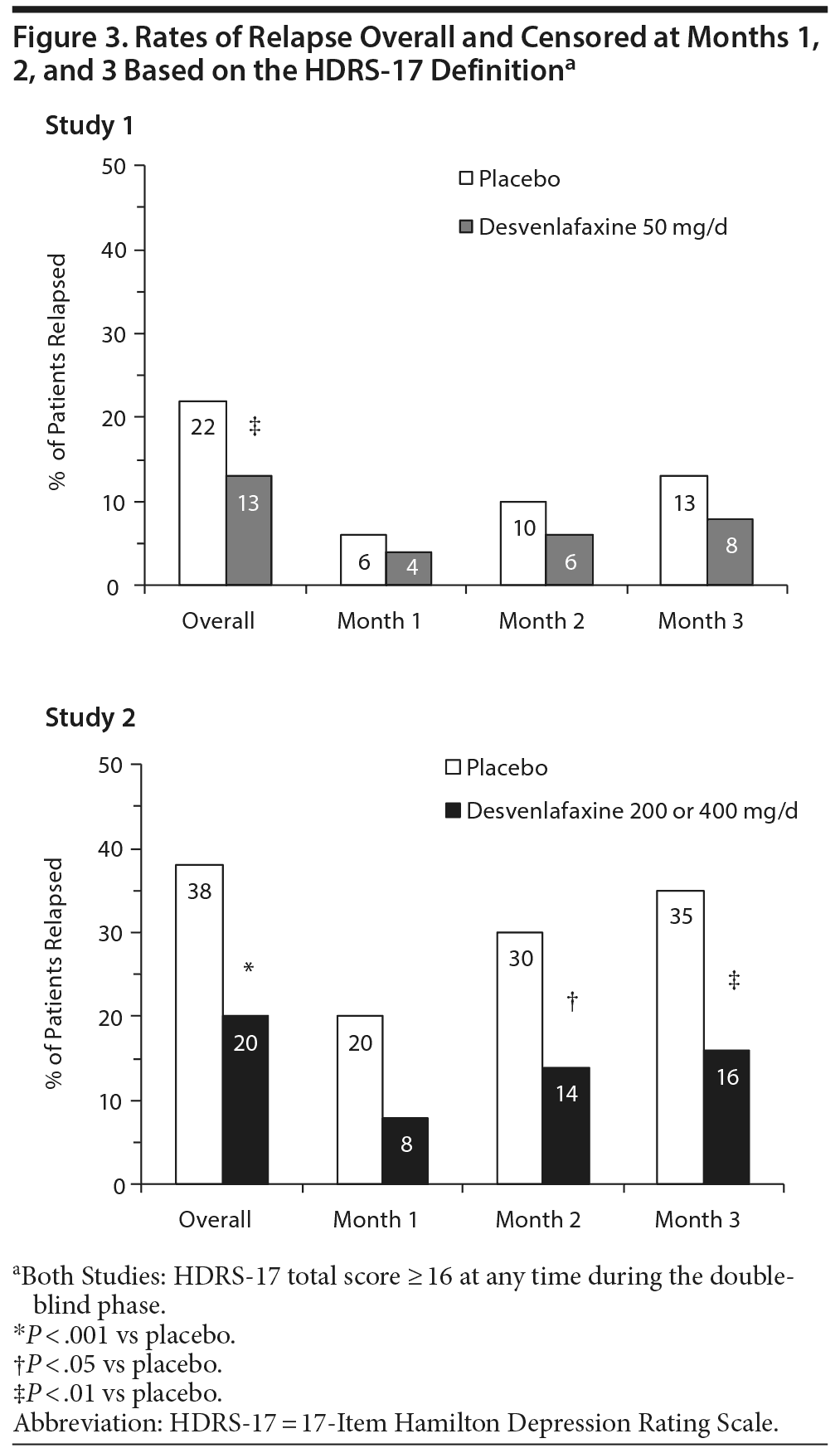
Hazard ratios for time to relapse and estimated probability of relapse censored at months 1, 2, and 3 and overall (month 6) for both studies are presented in Table 2. In study 1, 29% of placebo-treated patients and 35% of desvenlafaxine-treated patients who relapsed, based on the protocol definition, did so during the first month of the double-blind period, and relapse rates were significantly lower for desvenlafaxine versus placebo, censoring data at months 2 (P = .016) and 3 (P = .007), but not at month 1 (P = .088). Using the HDRS-17 definition, relapse rates were 28% and 32%, respectively, in the first month, and were not significantly lower for desvenlafaxine versus placebo, censoring data at months 1, 2, or 3. In study 2, 54% of placebo-treated patients and 47% of desvenlafaxine-treated patients who relapsed, by the protocol definition, did so during the first month of the double-blind period (52% and 43%, respectively, by the HDRS-17 definition). Relapse rates were significantly lower for desvenlafaxine compared with placebo, censoring data at months 1, 2, or 3, using protocol or HDRS-17 definitions (all P ≤ .002).
Predictors of Relapse
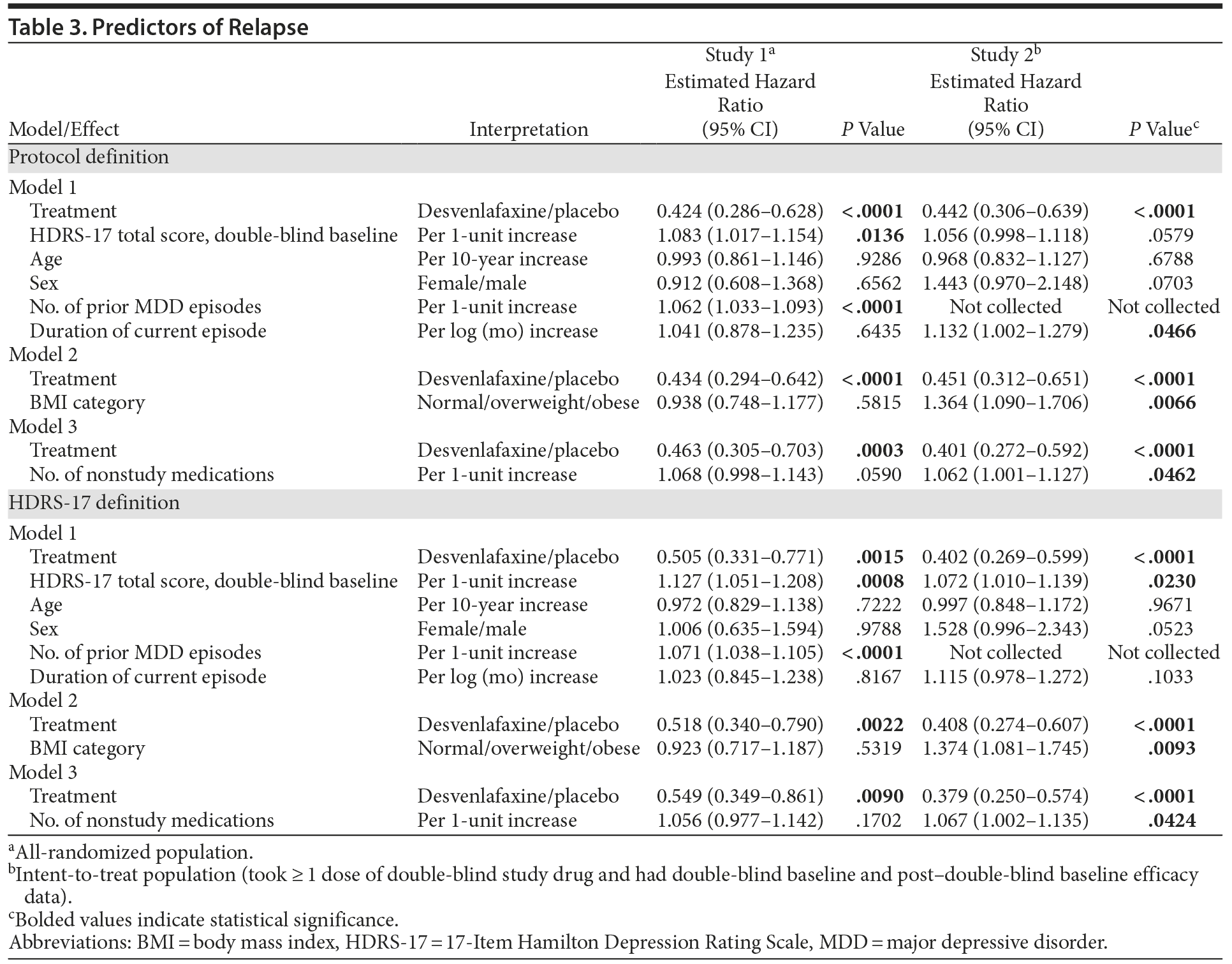
Results of the Cox proportional hazards models were similar for the 2 definitions of relapse (Table 3). Treatment was the only significant predictor of relapse for both studies using both definitions (all P ≤ .0015); higher HDRS-17 total score at double-blind baseline was a significant predictor of relapse for both studies using the HDRS-17 definition (both P ≤ .0230). Other statistically significant predictors of relapse for at least 1 of the studies for both definitions of relapse included number of prior MDD episodes (collected in study 1 only; both P < .0001), BMI (study 2, both P ≤ .0093), and number of nonstudy medications (study 2, both P ≤ .0462). In the analysis based on the protocol definitions, duration of current MDD episode was also a significant predictor of relapse in study 2 (P = .0466).
DISCUSSION
The duration of treatment prior to the randomized withdrawal phase varies across studies and may be a contributing factor in the differences observed in rates of relapse for both active and placebo groups. A 2-phase design, like study 218 in the current analysis, has been used in numerous relapse prevention studies, with the open-label phase duration commonly ranging from 6 weeks to 14 weeks.23–35 In this analysis, the ratio of the relapse rates for placebo compared with desvenlafaxine was 2 to 1 after both the 12-week open-label treatment in study 2 and the 20-week open-label treatment in study 1,17 but the rates for both treatment groups were numerically lower in the trial with the longer open-label treatment period. Other differences between the trials may have contributed to the lower observed relapse rates in study 1. More stringent entry criteria for the randomized withdrawal phase were used in study 1 compared with study 2 (HDRS-17 and CGI-I cutoffs versus HDRS-17 only), and patients assigned to double-blind treatment in study 1 met criteria at 2 different time points, demonstrating a stable response over at least 12 weeks of open-label treatment. Nonetheless, study 1 is the first conducted to date that assessed rates of relapse of depression using a longer stabilization phase design, and the difference in observed relapse rates between these 2 studies suggests that longer treatment durations may be beneficial in future antidepressant studies.
The observed timing of relapse also differed in studies 1 and 2. Of those who relapsed during the double-blind period, a greater proportion of patients relapsed early (month 1) after the 12-week open-label phase in study 2 (placebo: 54%; desvenlafaxine: 47%) compared with after the 20-week open-label phase in study 1 (placebo: 29%; desvenlafaxine: 35%). The difference between desvenlafaxine and placebo was statistically significant for data censored at month 1 in study 2 but not in study 1. It is possible that the month 1 treatment difference in the relapse rate was exaggerated in study 2 if withdrawal symptoms related to the rapid taper from desvenlafaxine 200 or 400 mg/d to placebo after randomization were mistaken for relapse symptoms. Nonetheless, approximately 80% to 90% of patients in either treatment group who relapsed in study 2 (either definition) did so during the first 3 months of the double-blind period, whereas over a third (35%–42%) of the relapses observed in study 1 occurred after the third month of the double-blind period. Randomized withdrawal trials in the literature using a longer 3-phase design similar to that of study 1 were designed to assess the prevention of recurrence of depression.36–38 It may be that, for some patients in study 1, meeting the relapse criteria months after demonstrating a stable response to treatment signaled a recurrence of depression rather than relapse of the same depressive episode.39–41
Predictors of depressive relapse in studies 1 and 2 were consistent with published analyses of antidepressant relapse prevention trials or naturalistic studies.2,31,42–45 Treatment was the only significant predictor of relapse for the 2 studies using HDRS-17 and protocol definitions of relapse. Using the HDRS-17 definition, residual symptoms after response to treatment—assessed by HDRS-17 total score at double-blind baseline—were also a significant predictor of relapse for both studies. Residual symptoms and the number of previous MDD episodes, which was a significant predictor in the 1 trial it was collected (study 1), are among the most commonly reported predictors of relapse of a depressive episode in published analyses.2,31,42–45
Conclusions about the underlying causes of differences in rates and timing of relapse between the 2 studies analyzed here are limited by the multiple differences in study design, including duration of the open-label period, differences in the definition of relapse, and 2 levels of response criteria in study 1 (response and stability) compared with response only in study 2. In addition, studies 1 and 2 used different desvenlafaxine dosing; study 2 used flexible dosing (200 or 400 mg/d) during the open-label period and 2 desvenlafaxine dose arms in the double-blind period, whereas study 1 used a single desvenlafaxine dose (50 mg/d) in both open-label and double-blind periods. Due to the differences between the 2 studies, they could not be compared statistically, and the analysis was limited to a side-by-side comparison. The generalizability of the findings is also limited by the source of the sample populations and enrollment criteria. Patients recruited at psychiatric practices and research centers may differ from those seen in primary care practices. In addition, the enrollment criteria for these studies were designed to select generally healthy patients with a primary diagnosis of MDD who therefore may differ from the broader population of depressed patients treated in clinical practice.
CONCLUSIONS
Desvenlafaxine 50 mg/d and desvenlafaxine 200 mg/d or 400 mg/d effectively reduced probability of relapse and extended time to relapse at 6 months compared with placebo. Relapse rates were numerically lower for both desvenlafaxine and placebo groups in study 1, which used a 20-week open-label period (response and stability phases), compared with study 2, which used a 12-week open-label response phase only. Early (month 1) relapse rates were statistically significantly lower for desvenlafaxine versus placebo only in study 2. These results could be explained in part by different study designs, including differences in duration of open-label stabilization.
Drug names: desvenlafaxine (Pristiq), venlafaxine (Effexor and others).
Author affiliations: Department of Psychiatry and Pharmacology, University of Toronto, Toronto, Ontario, Canada (Dr McIntyre); Pfizer Inc, New York, New York (Dr Fayyad); CGP Strategic Solutions LLC Lansdale, Pennsylvania (Dr Guico-Pabia); and Pfizer Canada, Inc, Kirkland, Quebec, Canada (Dr Boucher).
Potential conflicts of interest: Dr McIntyre has participated on Advisory Boards with the following companies: Astra Zeneca, Bristol-Myers Squibb, France Foundation, GlaxoSmithKline, Janssen-Ortho, Eli Lilly, Organon, Lundbeck, Pfizer, Shire, and Merck. He has participated in Speakers Bureaus with Janssen-Ortho, AstraZeneca, Eli Lilly, Lundbeck, Merck, and Pfizer. He participates in CME activities with AstraZeneca, Bristol-Myers Squibb, France Foundation, I3CME, Physicians Postgraduate Press, CME Outfitters, Optum Health, Merck, Eli Lilly, and Pfizer. He also received research grants from Eli Lilly, Janssen-Ortho, Shire, AstraZeneca, Pfizer, and Lundbeck. Drs Boucher and Fayyad are current Pfizer employees and shareholders. Dr Guico-Pabia was an employee and shareholder of Pfizer at the time the analysis was completed.
Funding/support: This analysis was sponsored by Pfizer. The original studies were sponsored by Wyeth Research, which was acquired by Pfizer in October 2009. No author received an honorarium or other form of financial support related to the development of this manuscript.
Role of the sponsor: The sponsor contributed to the design and conduct of the study; collection and analysis of data; and review of the manuscript. The authors were responsible for the interpretation of the data, the final content of the manuscript, and the decision to submit for publication.
Previous presentation: Data in this manuscript were presented at the annual meeting of the American Psychiatric Association; May 18–23, 2013; San Francisco, California.
Acknowledgments: Medical writing support was provided by Kathleen Dorries, PhD, of Peloton Advantage and was funded by Pfizer. Dr Dorries reports no other conflicts of interest related to the subject of this article. The authors would like to thank the patients who participated in studies 1 and 2 and the study site personnel, who were essential to the conduct of the studies.
REFERENCES
1. Judd LL, Akiskal HS, Maser JD, et al. Major depressive disorder: a prospective study of residual subthreshold depressive symptoms as predictor of rapid relapse. J Affect Disord. 1998;50(2–3):97–108. doi:10.1016/S0165-0327(98)00138-4 PubMed
2. Nierenberg AA, Husain MM, Trivedi MH, et al. Residual symptoms after remission of major depressive disorder with citalopram and risk of relapse: a STAR*D report. Psychol Med. 2010;40(1):41–50. doi:10.1017/S0033291709006011 PubMed
3. Glue P, Donovan MR, Kolluri S, et al. Meta-analysis of relapse prevention antidepressant trials in depressive disorders. Aust N Z J Psychiatry. 2010;44(8):697–705. doi:10.3109/00048671003705441 PubMed
4. Patten SB, Kennedy SH, Lam RW, et al; Canadian Network for Mood and Anxiety Treatments (CANMAT). Canadian Network for Mood and Anxiety Treatments (CANMAT) clinical guidelines for the management of major depressive disorder in adults, 1: classification, burden and principles of management. J Affect Disord. 2009;117(suppl 1):S5–S14. doi:10.1016/j.jad.2009.06.044 PubMed
5. Lam RW, Kennedy SH, Grigoriadis S, et al; Canadian Network for Mood and Anxiety Treatments (CANMAT). Canadian Network for Mood and Anxiety Treatments (CANMAT) clinical guidelines for the management of major depressive disorder in adults, 3: pharmacotherapy. J Affect Disord. 2009;117(suppl 1):S26–S43. doi:10.1016/j.jad.2009.06.041 PubMed
6. National Institute for Health and Clinical Excellence. Depression in adults: the treatment and management of depression in adults. NICE Web site. 2009. https://www.nice.org.uk/guidance/cg90. Accessed July 29, 2013.
7. Anderson IM, Ferrier IN, Baldwin RC, et al. Evidence-based guidelines for treating depressive disorders with antidepressants: a revision of the 2000 British Association for Psychopharmacology guidelines. J Psychopharmacol. 2008;22(4):343–396. doi:10.1177/0269881107088441 PubMed
8. Davidson JR. Major depressive disorder treatment guidelines in America and Europe. J Clin Psychiatry. 2010;71(suppl E1):e04. doi:10.4088/JCP.9058se1c.04gry PubMed
9. Work Group on Major Depressive Disorder. Practice guideline for the treatment of patients with major depressive disorder. American Psychiatric Association. 2010. http://www.psychiatryonline.com/pracGuide/pracGuideTopic_7.aspx. Accessed February 1, 2012.
10. Moore M, Yuen HM, Dunn N, et al. Explaining the rise in antidepressant prescribing: a descriptive study using the general practice research database. BMJ. 2009;339(2):b3999. doi:10.1136/bmj.b3999 PubMed
11. Lockhart P, Guthrie B. Trends in primary care antidepressant prescribing 1995–2007: a longitudinal population database analysis. Br J Gen Pract. 2011;61(590):e565–e572. doi:10.3399/bjgp11X593848 PubMed
12. Sawada N, Uchida H, Suzuki T, et al. Persistence and compliance to antidepressant treatment in patients with depression: a chart review. BMC Psychiatry. 2009;9(1):38. doi:10.1186/1471-244X-9-38 PubMed
13. Burton C, Anderson N, Wilde K, et al. Factors associated with duration of new antidepressant treatment: analysis of a large primary care database. Br J Gen Pract. 2012;62(595):e104–e112. doi:10.3399/bjgp12X625166 PubMed
14. Furukawa TA, Onishi Y, Hinotsu S, et al. Prescription patterns following first-line new generation antidepressants for depression in Japan: a naturalistic cohort study based on a large claims database. J Affect Disord. 2013;150(3):916–922. doi:10.1016/j.jad.2013.05.015 PubMed
15. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals, Inc, a subsidiary of Pfizer Inc; 2013.
16. Pristiq

17. Rosenthal JZ, Boyer P, Vialet C, et al. Efficacy and safety of desvenlafaxine 50 mg/d for prevention of relapse in major depressive disorder: a randomized controlled trial. J Clin Psychiatry. 2013;74(2):158–166. doi:10.4088/JCP.12m07974 PubMed
18. Rickels K, Montgomery SA, Tourian KA, et al. Desvenlafaxine for the prevention of relapse in major depressive disorder: results of a randomized trial. J Clin Psychopharmacol. 2010;30(1):18–24. doi:10.1097/JCP.0b013e3181c94c4d PubMed
19. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23(1):56–62. doi:10.1136/jnnp.23.1.56 PubMed
20. Guy W. Clinical Global Impressions. ECDEU Assessment Manual for Psychopharmacology. Rockville, MD: US Department of Health, Education, and Welfare; 1976:217–222.
21. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
22. Sheehan DV, Lecrubier Y, Sheehan KH, et al. The Mini-International Neuropsychiatric Interview (MINI): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59(suppl 20):22–33, quiz 34–57. PubMed
23. Montgomery SA, Rasmussen JG, Tanghøj P. A 24-week study of 20 mg citalopram, 40 mg citalopram, and placebo in the prevention of relapse of major depression. Int Clin Psychopharmacol. 1993;8(3):181–188. doi:10.1097/00004850-199300830-00008 PubMed
24. Entsuah AR, Rudolph RL, Hackett D, et al. Efficacy of venlafaxine and placebo during long-term treatment of depression: a pooled analysis of relapse rates. Int Clin Psychopharmacol. 1996;11(2):137–145. PubMed
25. Reimherr FW, Amsterdam JD, Quitkin FM, et al. Optimal length of continuation therapy in depression: a prospective assessment during long-term fluoxetine treatment. Am J Psychiatry. 1998;155(9):1247–1253. PubMed
26. Schmidt ME, Fava M, Robinson JM, et al. The efficacy and safety of a new enteric-coated formulation of fluoxetine given once weekly during the continuation treatment of major depressive disorder. J Clin Psychiatry. 2000;61(11):851–857. doi:10.4088/JCP.v61n1107 PubMed
27. Joliat MJ, Schmidt ME, Fava M, et al. Long-term treatment outcomes of depression with associated anxiety: efficacy of continuation treatment with fluoxetine. J Clin Psychiatry. 2004;65(3):373–378. doi:10.4088/JCP.v65n0313 PubMed
28. Simon JS, Aguiar LM, Kunz NR, et al. Extended-release venlafaxine in relapse prevention for patients with major depressive disorder. J Psychiatr Res. 2004;38(3):249–257. doi:10.1016/j.jpsychires.2003.10.004 PubMed
29. Rapaport MH, Bose A, Zheng H. Escitalopram continuation treatment prevents relapse of depressive episodes. J Clin Psychiatry. 2004;65(1):44–49. doi:10.4088/JCP.v65n0107 PubMed
30. Fava M, Detke MJ, Balestrieri M, et al. Management of depression relapse: reinitiation of duloxetine treatment or dose increase. J Psychiatr Res. 2006;40(4):328–336. doi:10.1016/j.jpsychires.2005.06.005 PubMed
31. McGrath PJ, Stewart JW, Quitkin FM, et al. Predictors of relapse in a prospective study of fluoxetine treatment of major depression. Am J Psychiatry. 2006;163(9):1542–1548. doi:10.1176/appi.ajp.163.9.1542 PubMed
32. Perahia DG, Gilaberte I, Wang F, et al. Duloxetine in the prevention of relapse of major depressive disorder: double-blind placebo-controlled study. Br J Psychiatry. 2006;188(4):346–353. doi:10.1192/bjp.188.4.346 PubMed
33. Gorwood P, Weiller E, Lemming O, et al. Escitalopram prevents relapse in older patients with major depressive disorder. Am J Geriatr Psychiatry. 2007;15(7):581–593. doi:10.1097/01.JGP.0000240823.94522.4c PubMed
34. Boulenger JP, Loft H, Florea I. A randomized clinical study of Lu AA21004 in the prevention of relapse in patients with major depressive disorder. J Psychopharmacol. 2012;26(11):1408–1416. doi:10.1177/0269881112441866 PubMed
35. Goodwin GM, Boyer P, Emsley R, et al. Is it time to shift to better characterization of patients in trials assessing novel antidepressants? an example of two relapse prevention studies with agomelatine. Int Clin Psychopharmacol. 2013;28(1):20–28. doi:10.1097/YIC.0b013e32835b0814 PubMed
36. Montgomery SA, Entsuah R, Hackett D, et al; Venlafaxine 335 Study Group. Venlafaxine versus placebo in the preventive treatment of recurrent major depression. J Clin Psychiatry. 2004;65(3):328–336. doi:10.4088/JCP.v65n0307 PubMed
37. Gilaberte I, Montejo AL, de la Gandara J, et al; Fluoxetine Long-Term Study Group. Fluoxetine in the prevention of depressive recurrences: a double-blind study. J Clin Psychopharmacol. 2001;21(4):417–424. doi:10.1097/00004714-200108000-00009 PubMed
38. Kocsis JH, Thase ME, Trivedi MH, et al. Prevention of recurrent episodes of depression with venlafaxine ER in a 1-year maintenance phase from the PREVENT Study. J Clin Psychiatry. 2007;68(7):1014–1023. doi:10.4088/JCP.v68n0706 PubMed
39. Frank E, Prien RF, Jarrett RB, et al. Conceptualization and rationale for consensus definitions of terms in major depressive disorder: remission, recovery, relapse, and recurrence. Arch Gen Psychiatry. 1991;48(9):851–855. doi:10.1001/archpsyc.1991.01810330075011 PubMed
40. Kupfer DJ. Long-term treatment of depression. J Clin Psychiatry. 1991;52(suppl 52):28–34. PubMed
41. Nierenberg AA, DeCecco LM. Definitions of antidepressant treatment response, remission, nonresponse, partial response, and other relevant outcomes: a focus on treatment-resistant depression. J Clin Psychiatry. 2001;62(suppl 16):5–9. PubMed
42. Fava M, Wiltse C, Walker D, et al. Predictors of relapse in a study of duloxetine treatment in patients with major depressive disorder. J Affect Disord. 2009;113(3):263–271. doi:10.1016/j.jad.2008.05.023 PubMed
43. Lin EH, Katon WJ, VonKorff M, et al. Relapse of depression in primary care: rate and clinical predictors. Arch Fam Med. 1998;7(5):443–449. doi:10.1001/archfami.7.5.443 PubMed
44. Gopinath S, Katon WJ, Russo JE, et al. Clinical factors associated with relapse in primary care patients with chronic or recurrent depression. J Affect Disord. 2007;101(1–3):57–63. doi:10.1016/j.jad.2006.10.023 PubMed
45. Rush AJ, Wisniewski SR, Zisook S, et al. Is prior course of illness relevant to acute or longer-term outcomes in depressed outpatients? a STAR*D report. Psychol Med. 2012;42(6):1131–1149. PubMed
Please sign in or purchase this PDF for $40.00.
![]() Save
Save
![]() Share
Share
![]() Cite
Cite
