Objective: The purpose of this post hoc analysis was to evaluate the incidence and timing of taper/posttherapy-emergent adverse events (TPAEs) following discontinuation of long-term treatment with desvenlafaxine (administered as desvenlafaxine succinate).
Method: This was a phase 4, randomized, double-blind, placebo-controlled study conducted at 38 research centers within the United States between March 2010 and February 2011. Adult outpatients with major depressive disorder (MDD; DSM-IV-TR criteria) who completed 24 weeks of open-label treatment with desvenlafaxine 50 mg/d were randomly assigned to 1 of 3 groups for the double-blind taper phase: desvenlafaxine 50 mg/d for 4 weeks (no discontinuation), desvenlafaxine 25 mg/d for 1 week followed by placebo for 3 weeks (taper), or placebo for 4 weeks (abrupt discontinuation). The primary endpoint, Discontinuation-Emergent Signs and Symptoms Scale (DESS) score over the first 2 weeks of the taper phase, was described previously. Secondary assessments included incidence and timing of TPAEs (any adverse event that started or increased in severity during the double-blind phase) and the percentage of patients who could not continue the taper phase due to discontinuation symptoms. The Quick Inventory of Depressive Symptomatology Self-Report (QIDS-SR16) assessed MDD status.
Results: A total of 480 patients enrolled in the open-label phase; the full analysis set included 357 patients (taper, n = 139; abrupt discontinuation, n = 146; no discontinuation, n = 72). TPAEs occurred in all groups through week 4. The incidence of any TPAE was lower for taper versus abrupt discontinuation at week 1 (P < .001), similar at week 2, and lower for taper versus abrupt discontinuation at weeks 3 and 4 (P ≤ .034). The most common TPAEs (incidence ≥ 3%) in the taper group were nausea and headache (3% each) at week 1 and dizziness (5%) and headache (4%) at week 2. The most common TPAEs in the abrupt discontinuation group were dizziness (8%), headache (8%), nausea (4%), irritability (3%), and diarrhea (3%) at week 1 and headache (3%) at weeks 2 and 3. The most common TPAE in the no discontinuation group was nausea (6%) at week 2.
Conclusion: The overall incidence of any TPAE was lower in the taper versus abrupt discontinuation groups.
Trial Registration: ClinicalTrials.gov identifier: NCT01056289
Incidence and Timing of Taper/Posttherapy–Emergent
Adverse Events Following Discontinuation of Desvenlafaxine
50 mg/d in Patients With Major Depressive Disorder
ABSTRACT
Objective: The purpose of this post hoc analysis was to evaluate the incidence and timing of taper/posttherapy–emergent adverse events (TPAEs) following discontinuation of long-term treatment with desvenlafaxine (administered as desvenlafaxine succinate).
Method: This was a phase 4, randomized, double-blind, placebo-controlled study conducted at 38 research centers within the United States between March 2010 and February 2011. Adult outpatients with major depressive disorder (MDD; DSM-IV-TR criteria) who completed 24 weeks of open-label treatment with desvenlafaxine 50 mg/d were randomly assigned to 1 of 3 groups for the double-blind taper phase: desvenlafaxine 50 mg/d for 4 weeks (no discontinuation), desvenlafaxine 25 mg/d for 1 week followed by placebo for 3 weeks (taper), or placebo for 4 weeks (abrupt discontinuation). The primary endpoint, Discontinuation-Emergent Signs and Symptoms Scale (DESS) score over the first 2 weeks of the taper phase, was described previously. Secondary assessments included incidence and timing of TPAEs (any adverse event that started or increased in severity during the double-blind phase) and the percentage of patients who could not continue the taper phase due to discontinuation symptoms. The Quick Inventory of Depressive Symptomatology Self-Report (QIDS-SR16) assessed MDD status.
Results: A total of 480 patients enrolled in the open-label phase; the full analysis set included 357 patients (taper, n = 139; abrupt discontinuation, n = 146; no discontinuation, n = 72). TPAEs occurred in all groups through week 4. The incidence of any TPAE was lower for taper versus abrupt discontinuation at week 1 (P < .001), similar at week 2, and lower for taper versus abrupt discontinuation at weeks 3 and 4 (P ≤ .034). The most common TPAEs (incidence ≥ 3%) in the taper group were nausea and headache (3% each) at week 1 and dizziness (5%) and headache (4%) at week 2. The most common TPAEs in the abrupt discontinuation group were dizziness (8%), headache (8%), nausea (4%), irritability (3%), and diarrhea (3%) at week 1 and headache (3%) at weeks 2 and 3. The most common TPAE in the no discontinuation group was nausea (6%) at week 2.
Conclusion: The overall incidence of any TPAE was lower in the taper versus abrupt discontinuation groups.
Trial Registration: ClinicalTrials.gov identifier: NCT01056289
Prim Care Companion CNS Disord 2015;17(1):doi:10.4088/PCC.14m01715
© Copyright 2015 Physicians Postgraduate Press, Inc.
Submitted: August 26, 2014; accepted October 6, 2014.
Published online: February 5, 2015.
Corresponding author: Philip T. Ninan, MD, 21 W Main St, Washington, NC 27889 ([email protected]).
A common tolerability issue associated with antidepressant treatment of major depressive disorder (MDD) is the emergence of adverse events upon discontinuation of therapy.1,2 Discontinuation symptoms associated with the selective serotonin reuptake inhibitor (SSRI) and serotonin-norepinephrine reuptake inhibitor (SNRI) antidepressants include lethargy, dizziness, nausea, vomiting, headache, diarrhea, insomnia, anxiety, irritability, and agitation.1,3–5 These symptoms usually emerge within days of stopping antidepressant treatment6 and often resolve within weeks.2 However, for some patients, discontinuation-emergent adverse events can be more pronounced, last for a prolonged period of time, and cause significant morbidity.2
Clinical practice guidelines for the management of patients with MDD recommend that clinicians educate patients who are taking antidepressant therapy of the need to taper their medication because of the risk for discontinuation-emergent symptoms with abrupt discontinuation.7,8 A taper duration of several weeks is recommended for most antidepressant pharmacotherapies, including SSRIs and SNRIs, in order to minimize this risk, and clinicians should monitor patients over several months during and after discontinuation of treatment for any symptoms that may arise. In the 2010 practice guidelines, collaborators for the Work Group on MDD stressed the problematic nature of discontinuation symptoms, indicating that disturbances in mood, energy, sleep, and appetite could be misinterpreted as (or potentially mask) symptoms of a depressive relapse.7
The SNRI desvenlafaxine (administered as desvenlafaxine succinate) is approved by the US Food and Drug Administration for the treatment of adults with MDD.9 Desvenlafaxine has demonstrated efficacy for treating MDD at the recommended dose of 50 mg/d.10 In a pooled analysis of 10 desvenlafaxine clinical trials (dose range, 50–400 mg/d), discontinuation-emergent adverse events were reported by a greater percentage of patients with MDD who received desvenlafaxine compared with those who received placebo (40% vs 27%, respectively).11 In these studies, a majority of patients who received desvenlafaxine doses greater than 50 mg/d had the dose tapered over a 1- or 2-week period before discontinuing treatment; however, the desvenlafaxine 50-mg/d dose was not tapered. Thus, limited clinical data are available regarding the nature and incidence of discontinuation-emergent adverse events associated with discontinuation of desvenlafaxine 50 mg/d.
A phase 4, randomized, double-blind, placebo-controlled study evaluated the tolerability at discontinuation of long-term treatment with desvenlafaxine 50 mg/d for MDD using a taper regimen compared with abrupt discontinuation.12 The primary objective of this study, which was described in a separate publication,12 was to determine whether the occurrence of discontinuation symptoms, as measured by the Discontinuation-Emergent Signs and Symptoms Scale13 (DESS) total score, was equivalent for abrupt discontinuation versus a 1-week taper to desvenlafaxine 25 mg/d following 24 weeks of open-label treatment with desvenlafaxine 50 mg/d. The adjusted mean (SE) DESS total score over the first 2 weeks of the double-blind phase was 4.1 (0.72) for no discontinuation, 4.8 (0.54) for taper, and 5.3 (0.52) for abrupt discontinuation. The difference in adjusted mean DESS total scores between the abrupt-discontinuation and taper groups was 0.50, with a 95% confidence interval (CI) of –0.88 to 1.89, which was within the prespecified margin of equivalence (–2.5 to 2.5), therefore demonstrating equivalence between abrupt discontinuation of desvenlafaxine 50 mg/d and 1-week taper to desvenlafaxine 25 mg/d.
Secondary objectives of this phase 4 study, which are described herein, were to evaluate the incidence, nature, and timing of taper/posttherapy–emergent adverse events (TPAEs) following discontinuation of long-term treatment with desvenlafaxine 50 mg/d in patients with MDD, and to determine the percentage of patients who were unable to complete the double-blind taper phase because of discontinuation symptoms.
METHOD
This was a phase 4, randomized, double-blind, placebo-controlled study conducted at 38 research centers within the United States between March 2010 and February 2011 (ClinicalTrials.gov identifier: NCT01056289). The protocol and amendment received institutional review board or independent ethics committee approval, and the study was conducted in accordance with the International Conference on Harmonization Guideline for Good Clinical Practice and the ethical principles that have their origin in the Declaration of Helsinki. Written informed consent was obtained from all participants before any protocol-required procedures were performed.
Study Patients
Eligible participants included adult outpatients (≥ 18 years of age) with a primary diagnosis of single or recurrent MDD without psychotic features consistent with criteria from the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision.14 Patients were required to have depressive symptoms for ≥ 30 days prior to the screening visit and a 17-item Hamilton Depression Rating Scale15 (HDRS-17) total score ≥ 14 at baseline. Patients were excluded if they had a current primary diagnosis of anxiety disorder, significant risk of suicide based on the Columbia Suicide Severity Rating Scale16 at screening or baseline, current psychoactive substance abuse or dependence, clinically important medical illness, clinically important abnormalities on physical or laboratory evaluation, or history of seizure disorder, gastrointestinal disease, neoplastic disorder, or narrow-angle glaucoma.
Study Design
All eligible patients who received treatment with desvenlafaxine 50 mg/d for 24 weeks during the open-label phase were then eligible to enter a 4-week double-blind phase. Eligible patients were randomly assigned in a 1:2:2 ratio to (1) desvenlafaxine 50 mg/d for 4 weeks (no discontinuation), (2) desvenlafaxine 25 mg/d for 1 week followed by placebo for 3 weeks (taper), or (3) placebo for 4 weeks (abrupt discontinuation). All patients received a follow-up call at 1 and 2 weeks after the final dose of double-blind treatment.
Outcome Measures
The primary endpoint of the study, the DESS scale total score over the first 2 weeks of the double-blind phase, was described previously.12 The DESS is an observer-administered, 43-item assessment of discontinuation symptoms that was administered by medically qualified personnel for the purpose of assessing symptoms associated with discontinuing desvenlafaxine.
Secondary endpoints included the incidence and timing of TPAEs during the double-blind phase, and the percentage of patients who did not successfully complete the double-blind phase because of discontinuation symptoms. A TPAE was defined as any adverse event that started or increased in severity during the double-blind phase. The Quick Inventory of Depressive Symptomatology Self-Report (QIDS-SR16)17 was used to assess current MDD status. The QIDS-SR16 is a self-administered rating scale that assesses 16 items associated with depression. The HDRS-17 was assessed at study entry to determine eligibility, but was not assessed at any time point after screening. The incidence of TPAEs and serious adverse events was described previously.12
Exploratory endpoints included the Discontinuation Symptoms Severity Index (DSSI),12 an exploratory scale rating DESS items’ severity and relationship to discontinuation, and DESS score at the end of weeks 3 and 4 of the double-blind phase to determine if symptoms were present or worsened. Using the DSSI, for each new or worsened symptom noted on the DESS, information was obtained on the severity (mild, moderate, severe) and relationship to discontinuation (none/remote, possible, probable, definite). A new symptom was any symptom that appeared within 7 days before administration of the DESS, and an old symptom was any that emerged within the same timeframe but continued into the 7-day period. Old symptoms were classified as unchanged, improved, or worsened. The DSSI was administered by a trained clinician immediately following the DESS for any new or worsened symptom.
Statistical Analysis
The safety population included all patients who took ≥ 1 dose of study medication and were randomized into the double-blind phase. The full analysis set was defined as all randomized patients with ≥ 1 postrandomization DESS record. All statistical testing was done at the α ≤ .05, 2-sided level, and no adjustments were made for multiple comparisons. Demographic and baseline clinical characteristics were summarized in the safety population, and TPAEs were summarized in the full analysis set. Missing data were handled using the last observation carried forward approach (LOCF).
The percentage of patients with at least 1 TPAE was summarized by treatment group and weekly during the double-blind phase. Comparisons between treatment groups were done at each week using Fisher exact test. The incidence of the most common TPAEs (defined as incidence ≥ 2%) and the percentage of patients who were unable to complete the double-blind phase (ie, tapering of study medication) because of discontinuation symptoms were also summarized. QIDS-SR16 scores for the open-label and double-blind periods were summarized descriptively for all patients who took at least 1 dose of study medication and had at least 1 follow-up QIDS-SR16 assessment using LOCF.
For the exploratory analyses, DESS items rated on the DSSI as having a “possible,” “probable,” or “definite” relationship to discontinuation were categorized as “related.” DESS scores for items considered related to discontinuation were summarized descriptively by treatment arm at double-blind baseline and weekly during the double-blind phase using LOCF. The incidence of moderate or severe DESS symptoms and the incidence of severe DESS symptoms were also summarized descriptively during the double-blind phase. Each DSSI symptom was summarized descriptively by severity, relationship to discontinuation, and treatment arm in the full analysis set using observed cases.
RESULTS
Patient Disposition and Baseline Demographics
A total of 480 patients were enrolled in the open-label phase; 361 patients were randomized in the double-blind phase and included in the safety population (taper, n = 140; abrupt discontinuation, n = 148; no discontinuation, n = 73). A total of 118 patients discontinued the open-label phase; reasons for discontinuation were described previously.12 One patient completed the open-label phase, but was not randomized to the double-blind phase of the study. Demographics and baseline clinical characteristics were similar among the 3 treatment groups in the safety population during the double-blind phase (Table 1). A total of 357 patients had ≥ 1 postrandomization DESS record and were included in the full analysis set (taper, n = 139; abrupt discontinuation, n = 146; no discontinuation, n = 72).
Secondary Endpoints
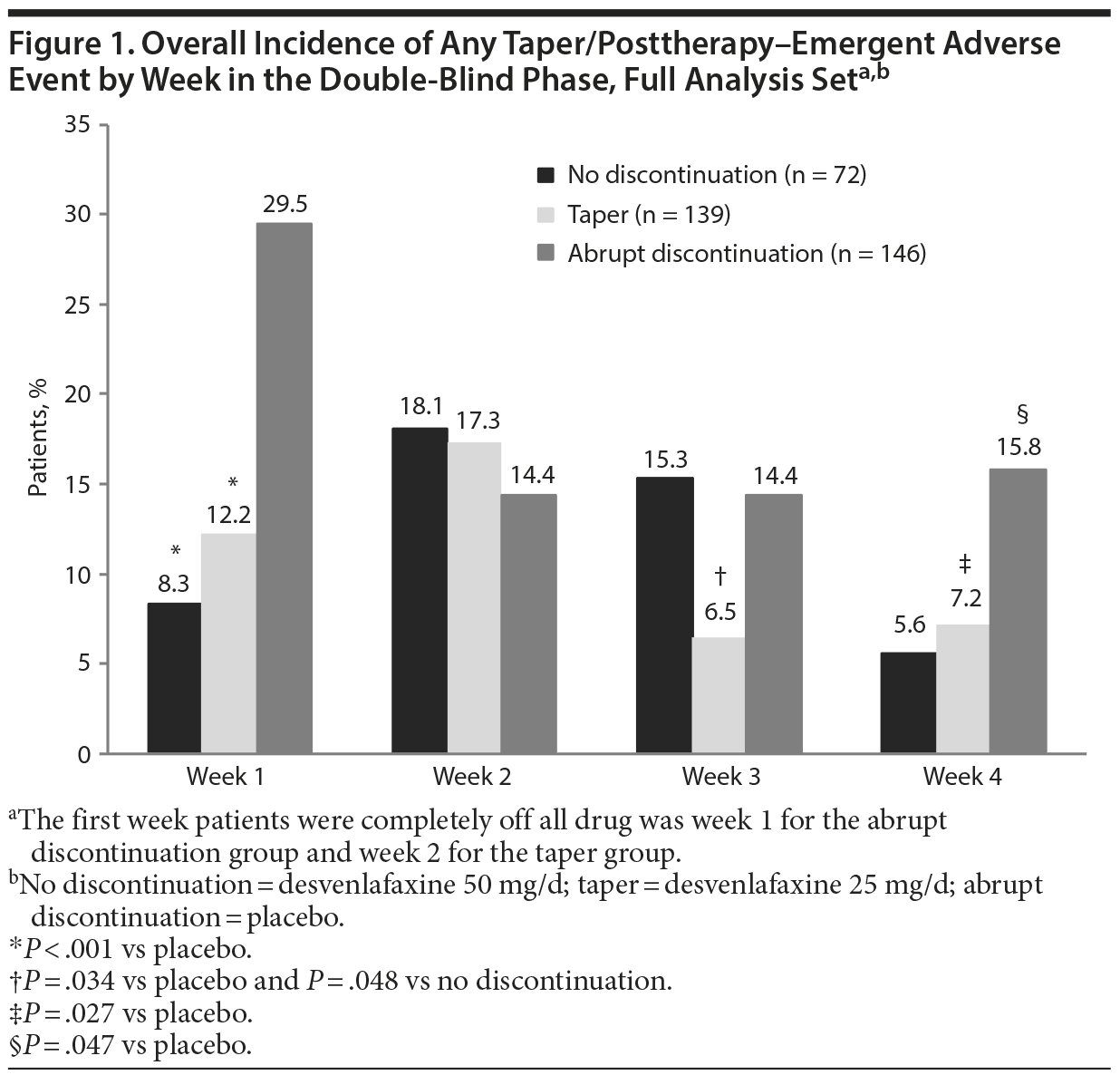
In the full analysis set, TPAEs were reported by 54 patients in the taper group (38.8%), 75 patients in the abrupt discontinuation group (51.4%), and 26 patients in the no discontinuation group (36.1%) during the double-blind phase.12 TPAEs occurred in all 3 groups through week 4, regardless of whether the desvenlafaxine dose was tapered (Figure 1). The overall incidence of any TPAE was significantly lower in the taper versus the abrupt discontinuation group at week 1 (P < .001), similar for the 2 groups at week 2 (P = .520), and significantly lower in the taper versus the abrupt discontinuation group at both weeks 3 (P = .034) and 4 (P = .027) (Figure 1). The overall incidence of any TPAE was highest in the abrupt discontinuation group at week 1 (29.5%), highest in the taper group at week 2 (17.3%), and highest in the no discontinuation group at week 2 (18.1%).
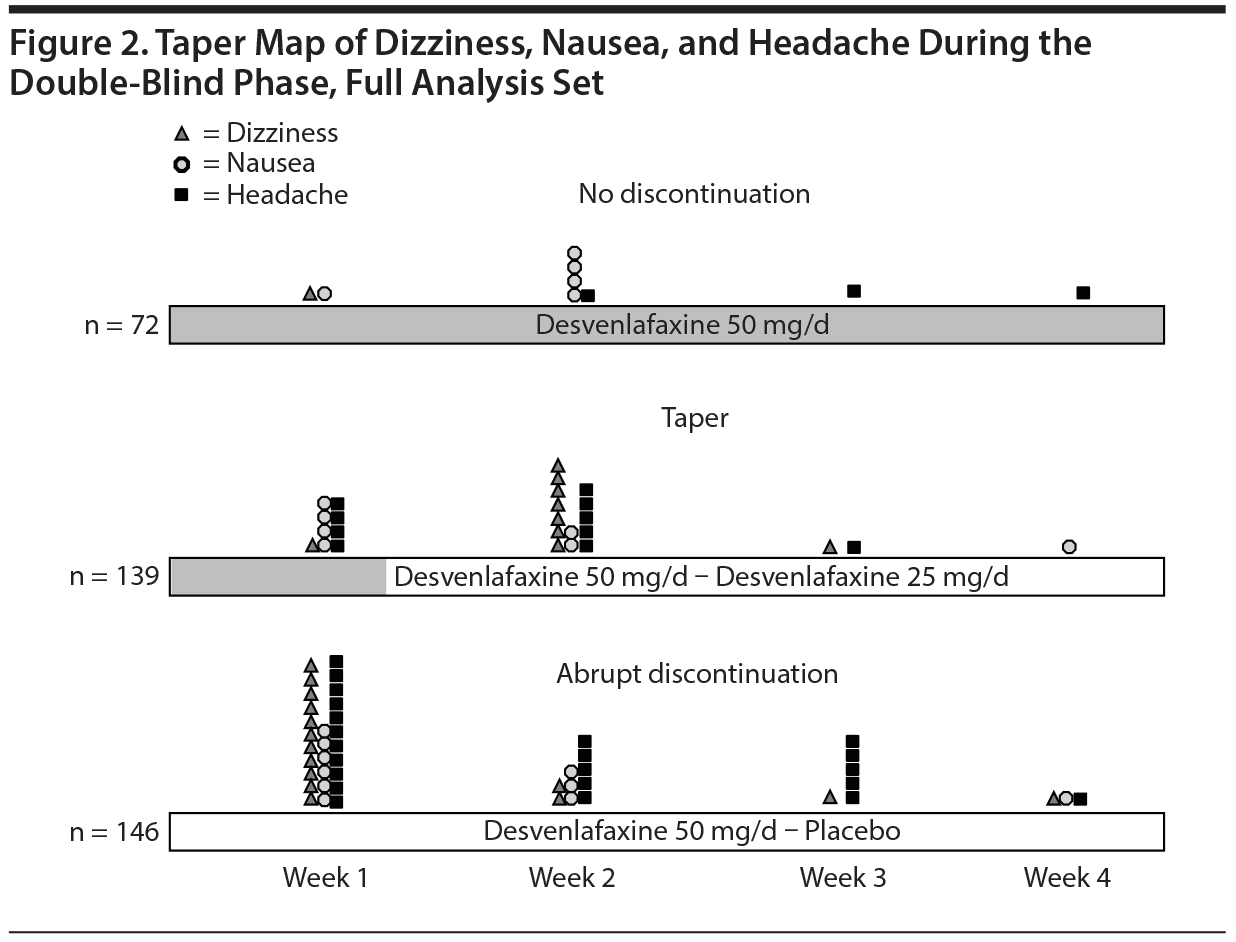
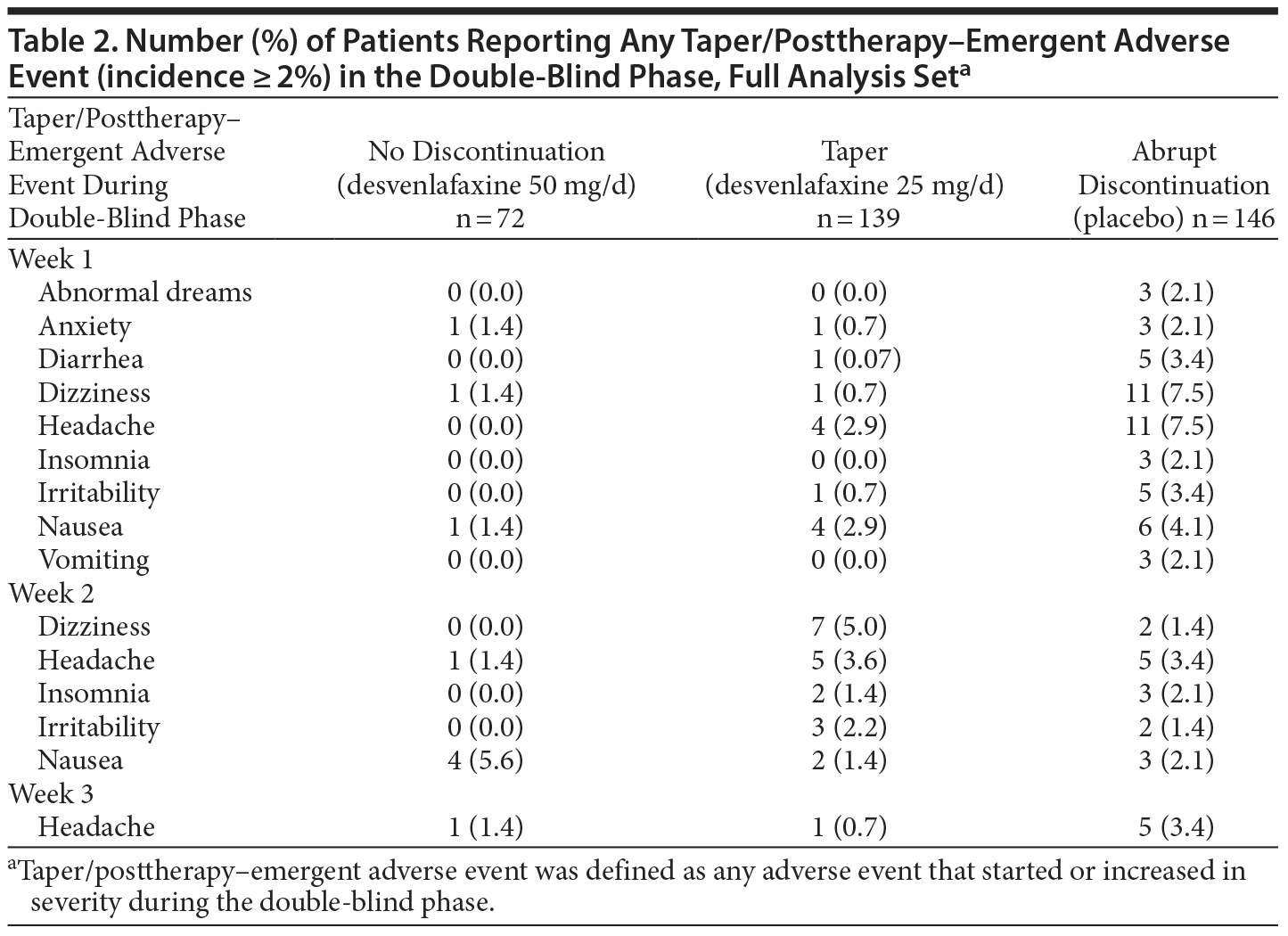
The 3 most common TPAEs overall were headache (9.1%), dizziness (6.4%), and nausea (5.5%). The timing of the 3 most common TPAEs is compared across groups in Figure 2. TPAEs with an incidence of 2% or more in any group are summarized by week in Table 2. A total of 5 patients did not successfully complete the double-blind phase because of discontinuation symptoms (no discontinuation group, n = 1 [1.4%]; taper group, n = 2 [1.4%]; abrupt discontinuation group, n = 2 [1.4%]).
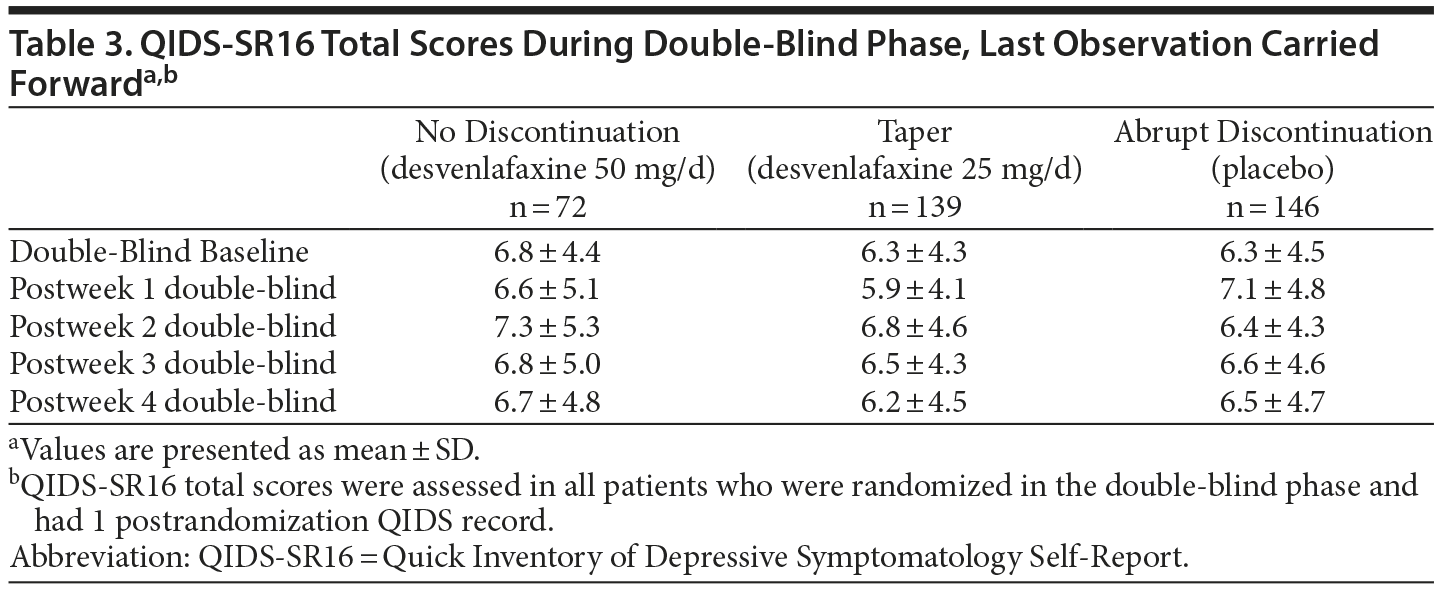
Total mean ± SD QIDS-SR16 scores decreased throughout the open-label phase from 13.0 ± 4.26 at open-label baseline to 4.3 ± 3.59 at 6 months (LOCF). No clinically significant changes in QIDS-SR16 scores were observed during the double-blind phase (Table 3).
Exploratory Endpoints
The mean number (SD) of DESS positive items considered related to discontinuation (based on DSSI rating of “possible,” “probable,” or “definite”) were lowest at baseline for all groups (taper: 0.0 [0.05]; abrupt discontinuation: 0.1 [0.33]; no discontinuation: 0.0 [0.07]). Scores were highest for the abrupt discontinuation group at post–double-blind week 1 (0.7 [1.57]) and for the taper group at post–double-blind week 2 (0.4 [0.86]). For the no discontinuation group, DESS scores for symptoms related to discontinuation were 0.3 (0.98) at post–double-blind week 1 and 0.3 (0.92) at post–double-blind week 2.There were no clinically significant differences among the treatment groups during the double-blind phase. During week 1 of the double-blind phase, which represented the first week off all active dose for the abrupt group and first week of taper, irritability was the most common DESS symptom related to discontinuation (7 patients in the no discontinuation group [9.7%], 22 patients in the taper group [15.8%], and 40 patients in the abrupt discontinuation group [27.4%]). Trouble sleeping/insomnia was the second most common DESS symptom related to discontinuation during week 1 (6 patients in the no discontinuation group [8.3%], 15 patients in the taper group [10.8%], and 32 patients in the abrupt discontinuation group [21.9%]).
During week 2 of the double-blind phase, which represented the first week off all active dose for the taper group, irritability was the most common DESS symptom related to discontinuation (2 patients in the no discontinuation group [2.8%], 27 patients in the taper group [19.4%], and 16 patients in the abrupt discontinuation group [11%]). Nervousness/anxiety was the second most common DESS symptom related to discontinuation during week 2 (8 patients in the no discontinuation group [11.1%], 18 patients in the taper group [12.9%], and 18 patients in the abrupt discontinuation group [12.3%]).
The majority of DESS symptoms were rated as mild or moderate in severity on the DSSI; the DESS symptom most frequently reported as severe was trouble sleeping/insomnia (3 patients in the no discontinuation group [4.2%], 3 patients in the taper group [2.2%], and 6 patients in the abrupt discontinuation group [4.1%]). At week 2, few patients had severe DESS symptoms as measured by the DSSI. Two patients in the no discontinuation group (2.8%), 1 patient in the taper group (0.7%), and 2 patients in the abrupt discontinuation group (1.4%) reported trouble sleeping/insomnia rated as severe on the DSSI. All other DESS symptoms rated as severe on the DSSI occurred in no more than 1 patient in any treatment group.
DISCUSSION
The purpose of this phase 4, randomized, double-blind, placebo-controlled study post hoc exploratory analysis was to assess the incidence and timing of TPAEs following discontinuation of long-term treatment with desvenlafaxine 50 mg/d in patients with MDD, and to determine the percentage of patients who were unable to complete the double-blind taper phase because of discontinuation symptoms. The primary analysis of this study has demonstrated that the number of discontinuation symptoms did not differ between taper and abrupt-discontinuation treatment groups based on the predetermined equivalence margin over the first and second week of the double-blind period.12 These post hoc analysis findings indicate that abrupt discontinuation of long-term treatment with desvenlafaxine 50 mg/d was equivalently tolerated compared with a 1-week taper to a desvenlafaxine 25-mg/d dose.
Overall, these findings support the primary analysis results. In the full analysis set (all randomized patients with ≥ 1 postrandomization DESS record), TPAEs occurred in all 3 groups through week 4, regardless of whether the desvenlafaxine dose was tapered. The overall incidence of any TPAE was significantly lower in the taper versus the abrupt discontinuation group at week 1, similar for the 2 groups at week 2, and significantly lower in the taper versus the abrupt discontinuation group at both weeks 3 and 4. The differences observed in timing of TPAEs were likely the result of the abrupt discontinuation and taper groups receiving their last dose of active drug at different times (week 1 and week 2, respectively). A greater incidence of TPAEs in week 1 for the abrupt discontinuation group and in week 2 for the taper group is consistent with what would be expected. In addition, the percentage of patients who did not successfully complete the double-blind taper phase because of discontinuation symptoms was the same across treatment groups (1.4% for each). Findings of the exploratory analysis showed that DESS scores related to discontinuation were not significantly different among the treatment groups during the double-blind taper phase, and most DESS symptoms were mild or moderate in severity.
The findings described herein support previously published reports of discontinuation symptoms associated with desvenlafaxine11 and other SNRIs.5,18,19 In a pooled analysis of short-term, fixed-dose trials of desvenlafaxine 50 mg/d to 400 mg/d in patients with MDD,11 dizziness, nausea, and headache were the most common discontinuation-emergent adverse events reported following cessation of treatment. In addition, DESS scores increased significantly compared with placebo after abrupt discontinuation of desvenlafaxine 50 mg/d. However, the analysis compared abrupt discontinuation with placebo and continuation of placebo, and not a taper regimen of desvenlafaxine.11
Few studies have made a direct comparison of tolerability between a taper regimen and abrupt discontinuation of SNRI treatment. One recent study20 was designed to assess the tolerability of 3 different taper regimens versus abrupt discontinuation following 15 weeks of open-label treatment with desvenlafaxine 100 mg/d for postmenopausal vasomotor symptoms. The 3 taper regimens studied were 1 week of desvenlafaxine 50 mg/d followed by 1 week of placebo, 1 week of desvenlafaxine 50 mg/d followed by 1 week of desvenlafaxine 25 mg/d, and 2 weeks of desvenlafaxine 50 mg/d every other day.20 No significant differences were observed between groups for either DESS scores or incidence of adverse events during the taper phase. However, the study explicitly excluded participants with MDD. Other studies examining the tolerability of SSRI or SNRI discontinuation have compared DESS scores at discontinuation between discontinuation versus no-discontinuation groups,21 between drug and placebo groups,22 or among different antidepressant drugs.13,19,21–23
While the results of this study show an occurrence of discontinuation symptoms regardless of discontinuation regimen, the lack of difference between the abrupt-discontinuation group and the taper group could be attributed to other factors, including blinding, randomization, or DESS administration. In addition, it is important to note that the patient population included generally healthy individuals with few significant medical or psychological comorbidities and taking limited concomitant medications. The treatment duration prior to discontinuation was 24 weeks. Thus, the overall generalizability to patients with MDD treated in clinical practice is limited by the exclusion criteria and duration of desvenlafaxine treatment. Finally, because no active comparator was included in the study, rates of discontinuation symptoms cannot be compared with those reported for other antidepressant pharmacotherapies. Nonetheless, these findings may help clinicians further educate patients of the expected nature and timing of potential discontinuation symptoms following cessation of treatment with desvenlafaxine.
CONCLUSIONS
Open-label treatment with desvenlafaxine 50 mg/d improved symptoms of MDD at 6 months compared with baseline, as indicated by improvements in QIDS-SR16 total scores. Tapering of desvenlafaxine 50 mg/d at 6 months did not eliminate the occurrence of discontinuation symptoms over the 4-week taper phase. In both the abrupt discontinuation and the taper groups, the percentage of patients who withdrew because of discontinuation symptoms was 1.4%. The most common TPAEs observed in the current study, including nausea, headache, and dizziness, are consistent with those reported in published reports of discontinuation symptoms associated with other SNRIs.
Drug names: desvenlafaxine (Pristiq).
Author affiliations: Brody School of Medicine, East Carolina University, Greenville, North Carolina (Dr Ninan); Pfizer Inc, Collegeville, Pennsylvania (Mr Musgnung and Ms Buckley); Pfizer Inc, New York, New York (Dr Messig); CGP Strategic Solutions, LLC, Lansdale, Pennsylvania (Dr Guico-Pabia); and National Institutes of Health, Rockville, Maryland (Dr Ramey).
Potential conflicts of interest: Drs Messig, Mr Musgnung, and Ms Buckley are employees of Pfizer. Dr Ninan is a former employee of and has received grant/research support from Pfizer. Dr Guico-Pabia is a a former employee of and a stock shareholder in Pfizer. Dr Ramey is a former employee of Pfizer and a stock shareholder in Eli Lilly and Pfizer. Drs Ninan, Guico-Pabia, and Ramey were employees of Pfizer at the time of the study.
Funding/support: This study was sponsored by Pfizer.
Role of the sponsor: The sponsor contributed to the design and conduct of the study, collection and analysis of data, and review of the manuscript. The authors were responsible for the interpretation of the data, the final content of the manuscript, and the decision to submit for publication.
Acknowledgments: Medical writing support was provided by Callie Grimes, PhD, of Peloton Advantage, LLC, Parsippany, New Jersey, and was funded by Pfizer.
Previous presentation: Poster presented at the American Psychiatric Association Annual Meeting; May 18–22, 2013; San Francisco, California.
REFERENCES
1. Warner CH, Bobo W, Warner C, et al. Antidepressant discontinuation syndrome. Am Fam Physician. 2006;74(3):449–456. PubMed
2. Haddad PM, Anderson IM. Recognising and managing antidepressant discontinuation symptoms. Adv Psychiatr Treat. 2007;13(6):447–457. doi:10.1192/apt.bp.105.001966
3. Black K, Shea C, Dursun S, et al. Selective serotonin reuptake inhibitor discontinuation syndrome: proposed diagnostic criteria. J Psychiatry Neurosci. 2000;25(3):255–261. PubMed
4. Haddad P. The SSRI discontinuation syndrome. J Psychopharmacol. 1998;12(3):305–313. doi:10.1177/026988119801200311 PubMed
5. Perahia DG, Kajdasz DK, Desaiah D, et al. Symptoms following abrupt discontinuation of duloxetine treatment in patients with major depressive disorder. J Affect Disord. 2005;89(1-3):207–212. doi:10.1016/j.jad.2005.09.003 PubMed
6. Bogetto F, Bellino S, Revello RB, et al. Discontinuation syndrome in dysthymic patients treated with selective serotonin reuptake inhibitors: a clinical investigation. CNS Drugs. 2002;16(4):273–283. doi:10.2165/00023210-200216040-00006 PubMed
7. Work Group on Major Depressive Disorder. Practice guideline for the treatment of patients with major depressive disorder. American Psychiatric Association. http://www.psychiatryonline.com/pracGuide/
pracGuideTopic_7.aspx. Accessed February 1, 2012.
8. National Institute for Clinical Excellence. Depression: Management of depression in primary and secondary care. Clinical guideline 23. National Institute for Clinical Excellence. http://www.nice.org.uk/guidance/cg023. Accessed February 1, 2012.
9. Pristiq [package insert]. Philadelphia, PA: Wyeth Pharmaceuticals, Inc, a subsidiary of Pfizer Inc; 2013.
10. Thase ME, Kornstein SG, Germain JM, et al. An integrated analysis of the efficacy of desvenlafaxine compared with placebo in patients with major depressive disorder. CNS Spectr. 2009;14(3):144–154. PubMed
11. Montgomery SA, Fava M, Padmanabhan SK, et al. Discontinuation symptoms and taper/poststudy-emergent adverse events with desvenlafaxine treatment for major depressive disorder. Int Clin Psychopharmacol. 2009;24(6):296–305. doi:10.1097/YIC.0b013e32832fbb5a PubMed
12. Khan A, Musgnung J, Ramey T, et al. Abrupt discontinuation compared with a 1-week taper regimen in depressed outpatients treated for 24 weeks with desvenlafaxine 50 mg/d. J Clin Psychopharmacol. 2014;34(3):365–368. PubMed
13. Rosenbaum JF, Fava M, Hoog SL, et al. Selective serotonin reuptake inhibitor discontinuation syndrome: a randomized clinical trial. Biol Psychiatry. 1998;44(2):77–87. doi:10.1016/S0006-3223(98)00126-7 PubMed
14. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision. Washington, DC: American Psychiatric Association; 2000.
15. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23(1):56–62. doi:10.1136/jnnp.23.1.56 PubMed
16. Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168(12):1266–1277. doi:10.1176/appi.ajp.2011.10111704 PubMed
17. Rush AJ, Trivedi MH, Ibrahim HM, et al. The 16-Item Quick Inventory of Depressive Symptomatology (QIDS), clinician rating (QIDS-C), and self-report (QIDS-SR): a psychometric evaluation in patients with chronic major depression. Biol Psychiatry. 2003;54(5):573–583. doi:10.1016/S0006-3223(02)01866-8 PubMed
18. Sir A, D’Souza RF, Uguz S, et al. Randomized trial of sertraline versus venlafaxine XR in major depression: efficacy and discontinuation symptoms. J Clin Psychiatry. 2005;66(10):1312–1320. doi:10.4088/JCP.v66n1015 PubMed
19. Montgomery SA, Huusom AK, Bothmer J. A randomized study comparing escitalopram with venlafaxine XR in primary care patients with major depressive disorder. Neuropsychobiology. 2004;50(1):57–64. doi:10.1159/000078225 PubMed
20. Gallagher JC, Strzinek RA, Cheng RF, et al. The effect of dose titration and dose tapering on the tolerability of desvenlafaxine in women with vasomotor symptoms associated with menopause. J Womens Health (Larchmt). 2012;21(2):188–198. doi:10.1089/jwh.2011.2764 PubMed
21. Montgomery SA, Kennedy SH, Burrows GD, et al. Absence of discontinuation symptoms with agomelatine and occurrence of discontinuation symptoms with paroxetine: a randomized, double-blind, placebo-controlled discontinuation study. Int Clin Psychopharmacol. 2004;19(5):271–280. doi:10.1097/01.yic.0000137184.64610.c8 PubMed
22. Baldwin DS, Montgomery SA, Nil R, et al. Discontinuation symptoms in depression and anxiety disorders. Int J Neuropsychopharmacol. 2007;10(1):73–84. doi:10.1017/S1461145705006358 PubMed
23. Baldwin DS, Cooper JA, Huusom AK, et al. A double-blind, randomized, parallel-group, flexible-dose study to evaluate the tolerability, efficacy and effects of treatment discontinuation with escitalopram and paroxetine in patients with major depressive disorder. Int Clin Psychopharmacol. 2006;21(3):159–169. doi:10.1097/01.yic.0000194377.88330.1d PubMed
Enjoy free PDF downloads as part of your membership!
![]() Save
Save
![]() Share
Share
![]() Cite
Cite
Advertisement
GAM ID: sidebar-top

